Diagnosis and management of inflammatory muscle disease
Idiopathic inflammatory myopathy is a term applied to a group of relatively rare diseases that present with the gradual onset of weakness of shoulder and pelvic girdle muscles. These diseases include polymyositis and dermatomyositis, as well as myositis associated with neoplastic disease, myositis associated with underlying collagen-vascular disease, and inclusion body myositis.
Weakness is a common clinical complaint that can be associated with significant morbidity. Although many complaints of weakness are purely subjective, the finding of objective muscle weakness indicates an underlying myopathic or neuropathic process. With an estimated incidence of only 5 cases per million adults,1the idiopathic inflammatory myopathies are rare diseases.
Individual diseases included under the term “idiopathic inflammatory myopathy” are polymyositis and dermatomyositis, as well as myositis associated with neoplastic disease, myositis associated with collagen-vascular disease, and inclusion body myositis. Although dermatomyositis can be seen in both adults and children, polymyositis is seen almost exclusively in adults. Inclusion body myositis is rare in patients younger than 60 years. Polymyositis and dermatomyositis are more common in women; inclusion body myositis is 3 times more common in men. Inclusion body myositis also is the most common idiopathic inflammatory myopathy. Myopathy associated with malignancy is more common in persons older than 50 years; myopathy associated with collagen-vascular disease occurs at the age that is associated with the particular collagen-vascular disease that is present.
The idiopathic inflammatory myopathies must be differentiated from those caused by infections, toxins, endocrinopathies, muscular dystrophies, and inborn errors of metabolism. Understanding the epidemiology, clinical presentation, diagnostic techniques, and treatment options is important to reduce the significant morbidities related to these disease processes. In this article, we describe an approach to the diagnosis and management of the idiopathic inflammatory myopathies.
DIAGNOSIS


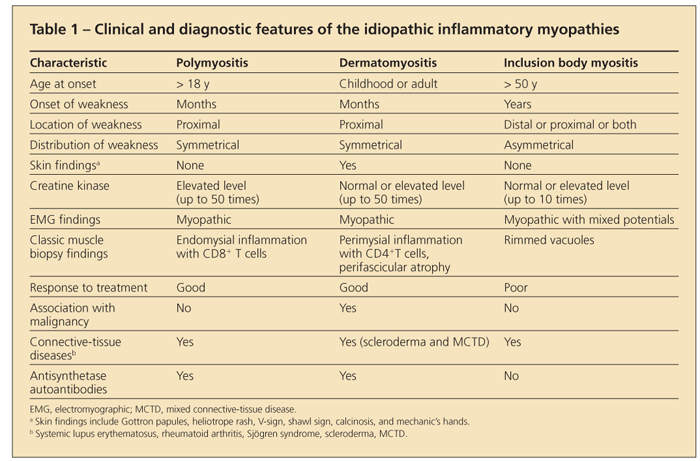
The diagnosis of an idiopathic inflammatory myopathy is made by finding a combination of a typical clinical presentation in association with elevated levels of enzymes of muscle origin, myopathic changes on electromyography, edema in muscle on MRI, and characteristic muscle histopathology. A summary of these clinical and diagnostic findings is shown in Table 1.
No single feature is diagnostic of an idiopathic inflammatory myopathy, and the diagnosis can be made only after the exclusion of other causes of these abnormalities. Although several sets of criteria have been developed to more accurately classify these diseases, none of them have been validated.2-4
History



Muscle weakness is the chief complaint in patients with any of the idiopathic inflammatory myopathies. However, muscle weakness is a common symptom in many patients with other conditions, and the differential diagnosis for the complaint is extensive (Table 2). A detailed history must be obtained and a thorough physical examination must be performed to rule out more common causes, such as prescription and illicit drug toxicities, environmental exposures, infections, and other signs or symptoms suggestive of endocrinopathies. A detailed family history may suggest the presence of an inherited muscle disorder, such as a muscular dystrophy or metabolic myopathy.
The first step in diagnosing an idiopathic inflammatory myopathy is to determine the time course of the weakness. Generally, weakness begins insidiously over a period of 3 to 6 months, without a recognized precipitating event. The weakness in patients with inclusion body myositis may develop even more insidiously over the course of years.
Other diseases that may cause the insidious onset of muscle weakness include the muscular dystrophies, metabolic myopathies (glycogen and lipid storage diseases), and endocrine myopathies. Neuropathic diseases, such as amyotrophic lateral sclerosis and myasthenia gravis, also may result in the gradual onset of weakness. The acute onset of muscle weakness suggests a neuropathic or myopathic cause other than the idiopathic inflammatory myopathies.
The second step is to determine the characteristics of the weakness and the effect it has on daily life. As a rule, myopathic weakness is symmetrical and occurs mostly in the proximal muscle groups of the pelvis, thighs, shoulders, and neck. For example, patients may complain of having difficulty in standing from a seated position, climbing stairs, brushing hair, or placing objects on a high shelf. Patients with inclusion body myositis provide an exception to the rule in that they may present with distal or asymmetrical muscle weakness.
The third step is to elicit any associated symptoms. In severe cases, esophageal and pharyngeal muscle weakness can result in dysphagia, aspiration pneumonia, and dysphonia. Extraocular muscles are spared. Less frequently seen are pulmonary (interstitial lung disease [ILD], diaphragmatic weakness) and cardiac manifestations (tachyarrhythmias, dilated cardiomyopathy). Muscle pain and joint pain may be seen but are not classic symptoms of the idiopathic inflammatory myopathies. Severe muscle pain and frank synovitis are uncommon. Because the idiopathic inflammatory myopathies can overlap with neoplastic disease and collagen-vascular diseases (system lupus erythematosus, Sjgren syndrome, scleroderma, and mixed connective-tissue disease [MCTD]), a detailed review of systems looking for symptoms of these conditions should be performed.
Physical examination
The physician should assess all muscle groups, noting that the weakness in most idiopathic inflammatory myopathies is proximal (shoulder, neck, and pelvic girdle muscle) and symmetrical. In contrast, neuropathic diseases typically present with distal or asymmetrical weakness and may present with abnormalities on neurological examination that would not be present in myopathic disease. However, many patients with inclusion body myositis may have asymmetrical weakness or weakness and atrophy of the distal muscle groups, providing a mixed picture.
The examiner should assess the patient's ability to swallow and speak as well as the patient's cardiac and pulmonary function, because these muscles and organs also may be involved. In addition, the physician must look for signs of neoplastic or other connective-tissue diseases.


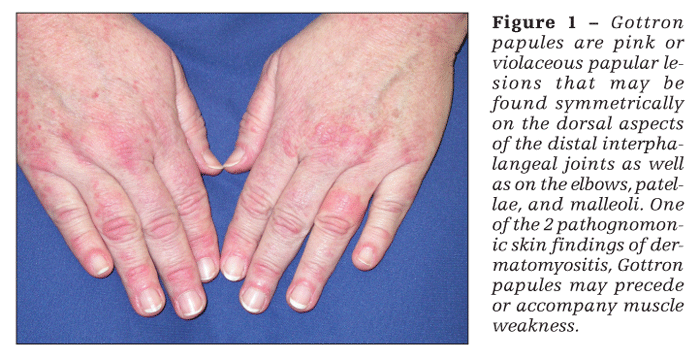
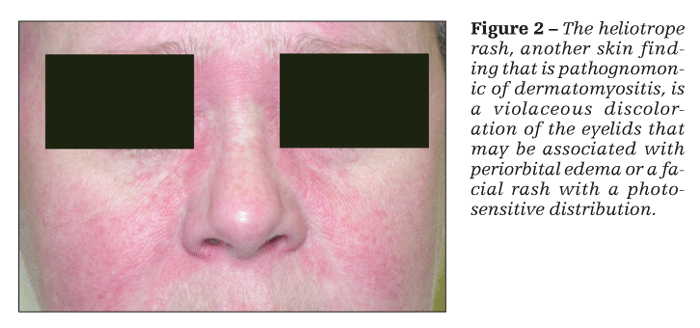
Patients with dermatomyositis have cutaneous findings that may precede or develop concomitantly with the muscle weakness or may develop after the onset of myopathy. The 2 pathognomonic skin findings of dermatomyositis are Gottron papules and the heliotrope rash. Gottron papules are pink or violaceous papular lesions that can be found symmetrically on the dorsal aspects of the distal interphalangeal joints, elbows, patellae, and malleoli (Figure 1). The heliotrope rash is a violaceous discoloration of the eyelids that may be associated with periorbital edema (Figure 2).
Other cutaneous findings in dermatomyositis include facial rash, the shawl sign, the V-sign, dystrophic cuticles, and mechanic's hands. Abnormal dilated capillaries may be seen just proximal to the cuticles. This finding is common in patients who also have Raynaud phenomenon.


Laboratory testing
Although serum elevations of aldolase, aspartate aminotransferase, alanine aminotransferase, and lactate dehydrogenase can be seen in myopathic disease, elevation of the creatine kinase (CK) level is thought to be the most sensitive laboratory test result related to skeletal muscle damage. However, the specificity of elevations in CK levels in regard to the idiopathic inflammatory myopathies is quite low. Severe disease may be seen with minimal elevations as well as with very high levels. In general, very high CK levels are seen more frequently as a result of medications, trauma, aerobic and isometric exercises, and other causes of muscle injury. In addition, there can be normal individual and racial variations in CK levels (highest levels in African American men), which may not be associated with a patient's complaint of weakness.5
Although elevated CK levels are seen in most cases of inflammatory myopathy, CK levels may be minimally elevated or even normal in inclusion body myositis, myositis associated with neoplasia, and early presentations of polymyositis and dermatomyositis and when significant muscle atrophy has developed. CK levels do not necessarily correlate with disease activity. Declines in CK levels may not accurately correlate with response to therapy, and disease remission can be attained despite persistent elevations in CK levels.
The erythrocyte sedimentation rate and C-reactive protein level often are normal in patients with myositis and therefore are not diagnostically helpful. Antinuclear antibodies (ANAs) often are present in low titers in patients with inflammatory myopathies. However, the presence of a high-titer ANA or of other positive serological test results may indicate the presence of myositis in association with another collagen-vascular disease, such as anti-Smith or anti–double-stranded DNA (system lupus erythematosus), anti-SSA(Ro) and anti-SSB(La) (Sjgren syndrome), anti-SCL-70 (scleroderma), and anti-ribonucleoprotein (MCTD).
Myositis-specific autoantibodies are found exclusively in patients with the idiopathic inflammatory myopathies. The most common of these antibodies are directed toward aminoacyl-tRNA synthetase activities. Patients who carry these specific antibodies may present with certain clinical features and may not respond well to treatment, depending on the type of antibody present.
Anti-Jo-1 is the most common of the antisynthetase antibodies; 73% of patients with anti-Jo-1 antibodies have ILD. These patients also are more likely to have fever, arthritis, Raynaud phenomenon, and mechanic's hands. This constellation of findings is referred to as the “antisynthetase syndrome.”6 Anti-Mi-2 antibodies are seen in up to 30% of patients with dermatomyositis and generally are associated with a very good prognosis. Anti–signal recognition particle (anti-SRP) antibodies are associated with cardiomyopathy and portend the worst prognosis.
Imaging
MRI is the most useful imaging modality for diagnosis and monitoring of myositis. MRI with T2-weighted images and fat suppression, or STIR technique, can show edema in the muscle, which is indicative of inflammation.7 Because there often is only patchy involvement of the muscle, MRI is an effective noninvasive method for determining the optimal location for muscle biopsy.
Electromyography
Electromyography is most useful for distinguishing between neuropathic and myopathic disease. Electromyographic (EMG) changes in the idiopathic inflammatory myopathies include the characteristic triad of (1) increased insertional activity, fibrillations, and sharp positive waves; (2) spontaneous, bizarre high-frequency discharges; and (3) polyphasic motor unit potentials of low amplitude and short duration.2 Although this triad is seen in 40% of patients with idiopathic inflammatory myopathies, 10% to 15% of patients will have normal EMG results.8 In patients with inclusion body myositis and distal or asymmetrical weakness, an EMG also may reveal some neuropathic changes.
Muscle biopsy
Muscle histology can help identify the specific type of idiopathic inflammatory myopathy; however, histological changes may be minimal or nonspecific. The classic findings of muscle inflammation are seen in polymyositis, dermatomyositis, and inclusion body myositis. Biopsy also may help with diagnosis of other myopathies, for which different treatment modalities would be required.
In polymyositis, muscle fibers are seen in various stages of necrosis and regeneration. T-cell infiltrates predominate, with CD8+ cytotoxic T cells most notably surrounding and invading muscle fibers. In later stages, muscle atrophy, fat, and fibrous connective tissue may be seen in the absence of inflammation.
Inflammation in dermatomyositis is predominantly perivascular and consists of CD4+ helper T cells, plasmacytoid dendritic cells, and B lymphocytes. Perifascicular atrophy is virtually diagnostic of dermatomyositis, regardless of whether inflammation is present. Thromboses of capillaries are also seen.
The histopathology of inclusion body myositis may be identical to that seen in polymyositis. If serial biopsies are performed, the degree of inflammatory change decreases and may disappear. The characteristic histopathological abnormality is intracellular lined (rimmed) vacuoles. Filamentous, intracytoplasmic, or intranuclear inclusions may be visualized by electron microscopy. Histochemical staining may show deposition of amyloid, ubiquitin, and SMI-31 in fibers.
Other testing
Dermatomyositis has a higher association with malignancies than polymyositis or inclusion body myositis. The most common cancers are those that occur most often in patients of a particular age and sex, with the exception of ovarian cancer, which is overexpressed.9
A CT scan of the chest, abdomen, and pelvis and a thorough screening for age-appropriate cancer are recommended at the time of diagnosis. Because of the association with ILD, patients with the anti-Jo-1 or other antisynthetase autoantibodies should receive baseline pulmonary function testing and a chest CT scan.
TREATMENT
Because the idiopathic inflammatory myopathies are relatively rare, referral to a specialist who is familiar with these diseases is recommended. Not only does medical treatment often require the use of immunosuppressive agents over long periods, but newer therapies and trials may be available that the nonspecialist may not be aware of.
Before medical therapy is started, a thorough assessment of a patient's individual muscle groups must be made so that his or her response to therapy may be measured objectively. Levels of CK and other muscle enzymes may be monitored during treatment; however, a decrease in these enzymes without any improvement in muscle weakness should not be mistaken for clinical improvement.10
Activity should be encouraged as soon as possible to prevent formation of muscle contractures. If weakness is severe, physical therapy should be prescribed to prevent flexion contractures. Otherwise, the patient should engage in regular aerobic and resistance exercise. Such exercise has been shown to not have adverse effects, and exercise has anti-inflammatory effects.11
Generally, the earlier in the disease course medical therapy is started, the more effective and rapid improvement in symptoms it produces. Empirically, corticosteroids are used as first-line therapy to decrease inflammation. In severe cases that require hospitalization, intravenous methylprednisolone may be used. Otherwise, prednisone may be started at 1 mg/kg/d. Ninety percent of patients who have polymyositis or dermatomyositis and are treated with corticosteroids achieve some clinical response; 50% to 75% achieve complete remission.
Risk factors for a poor outcome include concomitant ILD, significant esophageal dysmotility, cardiomyopathy, malignancy, and anti-SRP antibodies. However, corticosteroid myopathy, which can cause worsening muscle weakness, develops in some patients.
Prednisone should provide objective improvement in muscle weakness within 3 to 6 months. For patients who do not respond well to corticosteroids or those who present with more profound weakness, another immunosuppressive agent, such as methotrexate or azathioprine, is added next.12 Intravenous immunoglobulin may be used in severe disease to improve symptoms, but it is not effective in the long term or as a single agent.
Hydroxychloroquine, topical tacrolimus, and topical corticosteroids may be effective for cutaneous manifestations in patients with dermatomyositis13; however, they have no effect on the myositis. A recent systematic review highlighted the lack of quality studies assessing the efficacy and toxicity of various immunosuppressants in dermatomyositis and polymyositis.14 However, no medication has been found that alters the course of inclusion body myositis. Although weakness progresses in an unrelenting fashion, it does so very slowly and is not associated with increased mortality.
References:
References
1. Cronin ME, Plotz PH. Idiopathic inflammatory myopathies. Rheum Dis Clin North Am. 1990;16:655-665.
2. Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292:344-347.
3. Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med. 1975;292:403-407.
4. Mastaglia FL, Phillips BA. Idiopathic inflammatory myopathies: epidemiology, classification, and diagnostic criteria. Rheum Dis Clin North Am. 2002;28:723-741.
5. Black HR, Quallich H, Gareleck CB. Racial differences in serum creatine kinase levels. Am J Med. 1986;81:479-487.
6. Hengstman GJ, van Engelen BG, van Venrooij WJ. Myositis specific autoantibodies: changing insights in pathophysiology and clinical associations. Curr Opin Rheumatol. 2004;16:692-699.
7. Fraser DD, Frank JA, Dalakas M, et al. Magnetic resonance imaging in the idiopathic inflammatory myopathies. J Rheumatol. 1991;18:1693-1700.
8. Bohan A, Peter JB, Bowman RL, Pearson CM. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore). 1977;56:255-286.
9. Antiochos BB, Brown LA, Li Z, et al. Malignancy is associated with dermatomyositis but not polymyositis in northern New England, USA. J Rheumatol. 2009;36:2704-2710.
10. Kroll M, Otis J, Kagen L. Serum enzyme, myoglobin and muscle strength relationships in polymyositis and dermatomyositis. J Rheumatol. 1986;13:349-355.
11. Alexanderson H, Lundberg IE. The role of exercise in the rehabilitation of idiopathic inflammatory myopathies. Curr Opin Rheumatol. 2005;17:164-171.
12. Oddis CV. Idiopathic inflammatory myopathies. Chapter C: Treatment and assessment. In: Klippel JH, Stone JH, Crofford LJ, White PH, eds. Primer on the Rheumatic Diseases. 13th ed. Atlanta: Arthritis Foundation; 2008:375-380.
13. Callen JP, Wortmann RL. Dermatomyositis. Clin Dermatol. 2006;24:363-373.
14. Choy EH, Hoogendijk JE, Lecky B, Winer JB. Immunosuppressant and immunomodulatory treatment for dermatomyositis and polymyositis. Cochrane Database Syst Rev. 2005;(3):CD003643. Update in: Cochrane Database Syst Rev. 2009;(4):CD003643.














