Image IQ: Rash On the Chest with Muscle Weakness
A 51-year-old woman visits her primary care doctor to address a rash, pruritus, myalgias and muscle weakness. The rash was an erythematous blanching patch spread across her chest in a V shape. She had also noticed that her fingernail folds were red and swollen. What's your diagnosis?


Can you diagnose this patient? (©VisualDx)
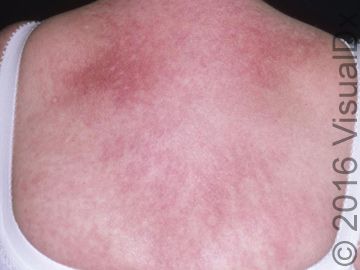
A 51-year-old woman visits her primary care doctor to address a rash, pruritus, myalgias and muscle weakness. The rash was an erythematous blanching patch spread across her chest in a V shape. She had also noticed that her fingernail folds were red and swollen.
Can you diagnose the patient? Use the differential builder in VisualDx as a guide.
A. Systemic lupus erythematosus
B. Polymorphous light eruption
C. Allergic contact dermatitis
D. Dermatomyositis
See the next page for the correct diagnosis.
ANSWER: D) Dermatomyositis
Synopsis
Dermatomyositis is a multisystem autoimmune connective tissue disease that is most often characterized by a symmetric proximal extensor inflammatory myopathy, a characteristic violaceous cutaneous eruption, and pathogenic circulating autoantibodies. Dermatomyositis demonstrates a bimodal incidence, with the adult form most commonly seen in individuals aged 45-60, and the juvenile form found in children aged 10-15 years. A 2:1 female-to-male incidence ratio exists in adults.
While the etiology remains unclear, some evidence suggests that genetically susceptible individuals with certain HLA types mount aberrant cellular and humoral responses after exposure to infection, malignancy, or drug ingestion.
Clinical features of dermatomyositis can be categorized into cutaneous and systemic manifestations. Typical findings include a heliotrope rash, atrophic dermal papules of dermatomyositis (ADPDM; previously Gottron's papules), shawl sign, holster sign, photosensitivity, flagellate erythema, poikiloderma, calcinosis cutis and nail fold changes. Pruritus is also common.
Systemic manifestations of dermatomyositis include fatigue, malaise, myalgias, and the following:
Musculoskeletal – Proximal extensor muscle group inflammation that leads to muscle pain and weakness. Patients may have difficulty getting up from a sitting position or combing their hair.
Gastrointestinal – Dysphagia can be seen due to esophageal muscle involvement.
Pulmonary – Some level of pulmonary involvement will be present in 15%-30% of patients. Diffuse interstitial fibrosis and acute respiratory distress syndrome are manifestations of pulmonary involvement.
Cardiac – Tachycardia or bradycardia, bundle branch blocks, cardiomegaly or congestive heart failure.
In addition, the clinician should be aware of several key points in patients with dermatomyositis:
Malignancy – Up to 40% of patients with the adult form may have an occult malignancy. Initial cancer screening and vigilant serial monitoring for 2-3 years post-diagnosis is strongly recommended. Chest, abdomen, and pelvis CT scanning – even in the absence of symptoms – should be considered. Commonly found malignancies include colon, ovarian, breast, pancreatic, lung, and gastric cancers and lymphoma. The juvenile form does not share this risk. The presence of anti-TIF1 IgG2 is a biomarker for increased risk (threefold) of malignancy.
Autoimmune disease overlap РDermatomyositis can occur in conjunction with systemic lupus erythematosus (SLE), mixed-connective tissue disease, Sj̦gren syndrome, scleroderma, and rheumatoid arthritis. Scleroderma / CREST syndrome (calcinosis cutis, Raynaud phenomena, esophageal dysfunction, sclerodactyly, and telangiectasia) is the most common overlap syndrome resulting in sclerodermatomyositis.
Antisynthetase syndrome – Fever, Raynaud phenomenon, "mechanic's hands," interstitial pulmonary fibrosis, polyarthritis, and autoantibodies to aminoacyl-tRNA synthetase.
Variant – Dermatomyositis sine myositis is the amyopathic form where dermatomyositis is clinically limited to cutaneous involvement. Disease is often detectable on muscle MRI, and many of these patients go on to develop muscle weakness as the disease progresses.
Variant – Anti-melanoma differentiation-associated protein (anti-MDA5) antibodies are found in up to 65% of clinically amyopathic dermatomyositis and in 10%-20% of patients with classic dermatomyositis. Their presence has been associated with the development of interstitial lung disease, rapidly progressive lung disease, and poor survival in both US and Asian studies. Anti-MDA5 antibodies have also been associated with mucocutaneous ulceration, palmar papules, nonscarring alopecia, panniculitis, and arthritis.
Dermatomyositis and Degos disease are, rarely, associated.
Dermatomyositis may be induced by medications, including hydroxyurea, penicillamine, interferon beta, and ipilimumab. Acute onset / flares of dermatomyositis have been reported in association with ingestion of IsaLean, an herbal supplement.
With proper therapy, 75% of patients can be disease free within 3 years.
For more information about this diagnosis and ICD 10 codes, visit VisualDx.














