Lung Disease in Scleroderma, Part I: Ten Points About ILD
Interstitial lung disease is a leading cause of mortality in systemic sclerosis. This quick ten-point review provides the latest and best information for diagnosis and management. (First of two parts.)


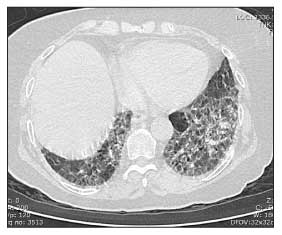
Recent literature in scleroderma (systemic sclerosis or SSc) has documented success in treating interstitial lung disease(ILD). This article discusses relevant questions about ILD in SSc including frequency, diagnosis and treatment.
This is the first of a two-part series. The second article will discuss pulmonary arterial hypertension (PAH) in systemic sclerosis.
How common is interstitial lung disease in systemic sclerosis (SSc, scleroderma)?
Also known as pulmonary fibrosis, ILD arises in approximately 1/3 of the diffuse cutaneous SSc subset and 1/4 of the limited cutaneous SSc subset.1 It is a leading cause of SSc-related mortality.2 Approximately half the patients with SSc die of their disease, and of these most die from ILD, followed by pulmonary arterial hypertension (PAH), cardiomyopathy, and renal crisis.2
ILD in SSc is clinically relevant (for instance, in 15% of patients the forced vital capacity [FVC] % predicted is less than 70%).3
Antitopoisomerase 1 (topo1) antibody, which is also called Scl70 antibody, is associated with an increased chance of having ILD in SSc. The most common type is bibasilar ILD with or without fibrosis (honey combing). It is usually NSIP (non-specific interstitial pneumonitis) and, far less often, usual interstitial pneumonitis (UIP).
How is SSc ILD diagnosed?
A detailed history of dyspnea is needed, because many patients deny shortness of breath as they accommodate to the slowly progressive pneumonitis. To determine whether a patient is actually symptomatic, it may help to ask about dyspnea in relation to specific activities (carrying parcels, walking outside, walking up a hill) or to ask comparative questions, such as ‘Compared to last year, has there been a change in your ability to do certain activities and if so, why?’ There are many dyspnea scales that can be used in research or clinical practice.4
All patients with SSc should have a chest x-ray. On physical examination, most patients show ausculation of bilateral basilar crackles (fine or coarse). A high resolution CT scan of the chest can rule in ILD and rule out other mimickers such as congestive heart failure.
Pulmonary function tests (PFTs) are done to determine the severity of the restrictive defect (i.e., low total lung capacity, forced vital capacity, residual volume and diffuse capacity).
Who should be treated for SSc ILD?
There is no consensus in the literature, but many would agree that patients who have ILD usually have the steepest rate of decline of PFTs in the first 5 years from their disease onset, and later die of ILD. They can remain stable for years and then deteriorate.
Treatment may be considered in cases of:
• moderate to severe ILD,
• recent deterioration of ILD,
• short duration of SSc with ILD, or
• presence of positive antitopoisomerase I (topo1) / Scl70 antibody.5
In an expert Delphi survey, most would treat symptomatic proven ILD and less than half would treat asymptomatic patients even if there was documented worsening of HRCT and/or PFTs.6 I would also consider treating those who have ILD in the presence of symptoms (dyspnea, severe cough).
How should SSc ILD be treated?
SSc ILD is treated with induction and maintenance therapy. Most experts would start with intravenous cyclophosphamide as induction, either either intravenous monthly pulse cyclophosphamide for 6 to 12 months or daily oral cyclophosphamide.6 Maintenance is with various immune suppressants. Three-fourths would consider mycophenylate mofetil (MMF) every three months for maintenance. Less often patients receive intravenous azathioprine or cyclophosphamide.6
The data for induction are based on two trials of cyclophosphamide in SSc ILD7,8 with the intravenous trial switching to maintenance with azathioprine.8 In a meta-analysis of the cyclophosphamide trials, pulmonary function appeared superior to placebo.9 If there is no maintenance therapy, the benefit is lost within a year or more after stopping cyclophosphamide. Patients regress and look similar to the placebo group.10
Guidelines from the European League Against Rheumatism (EULAR) recommend induction of ILD that is clinically relevant with cyclophosphamide and maintenance is needed, usually with a drug with a better safety profile.13
One small trial with induction using azathioprine compared to cyclophosphamide in SSc ILD demonstrated that azathioprine was not as effective.14What about treatment with other immune suppressants?
One single-site trial has shown effective induction with rituximab.11 Even after 2 years, the rituximab group had better FVC % predicted (73%) than the placebo group (68%).12 Another trial that was also positive would help to position rituximab as a more conventional treatment in these patients.
Pirfenidone (Esbriet), a small molecule inhibitor of TNF-α, has been approved for IPF, and studies in SSc ILD are underway.15What about steroids?
Steroids often are not effective as concomitant medication. If trying steroids, experts may use 1mg/kg/day of oral prednisone/prednisolone, rapidly tapering and discontinuing it if dyspnea has not improved after one month. If there is subjective improvement, the dose is lowered and maintained (if still effective) at 10 to 15mg/d. Steroids are only adjunctive treatment, and it is important to be aware of an increased risk of scleroderma renal crisis, especially in early diffuse SSc with high skin scores, tendon friction rubs, and male patients.6Other treatment in SSc ILD
Supportive treatment such as oxygen is recommended if there is desaturation present at rest or with exercise. Exercise may actually prolong life for some of these patients. Lung transplant for severe ILD in SSc has a survival curve that is initially better than that for idiopathic pulmonary fibrosis (IPF), but is similar at 5 years 16Is it important to prevent reflux with aspiration?
One large study of SSc patients with no ILD, stable ILD, or progressing ILD demonstrated that the largest factor in progression in a prevalent cohort was not topo1 antibody (which was associated with having ILD but not with ILD that progressed over the study period), but the occurrence of dysphagia requiring esophageal dilation as well as choking at night from acid reflux.1 The association was proven, but not a cause-and- effect relationship (as the data were from a prevalent cohort who were then followed prospectively). So treatment of aspiration and GERD and dysmotility could not be shown to decrease worsening ILD in that study. However, others have speculated that aspiration worsens SSc ILD, and that aggressive treatment of reflux and dysphagia may be helpful.17, 18What if ILD is concomitant with pulmonary arterial hypertension in SSc?
SSc patients who have a very low diffusing capacity (DLCO) % predicted, especially if it is far lower than the forced vital capacity (FVC)% predicted (such as a ratio of TLC % predicted/DLCO % predicted >2), should raise suspicion for concomitant pulmonary arterial hypertension.19 Approximately 15% of patients with ILD also have PAH, and the prognosis in these patients is very poor.2Are there other associations with ILD in SSc?
SSc patients with ILD have more digital ulcers than those without ILD. There is also an association between topo1 and digital ulcers or severe esophageal dysmotility.1, 20The take-home message:
• ILD is common in SSc.
• Approximately 15% of patients have clinically relevant ILD that often onsets early in the disease, with further decline in lung function later that contributes to increased mortality.
• There is not total consensus on who should be treated, but consider screening for ILD early in the disease and offering treatment with induction and then maintenance therapy especially for patients who show diffuse disease and /or test positive for topo1 antibody, The gold standard is induction with cyclophosphamide and then MMF or azthioprine for maintenance.
• All supportive care and treatment of gastroesophageal reflux should be considered in these patients.
• Ongoing research is seeking other drugs to treat ILD in SSc.
References:
REFERENCER
1. Zhang XJ, Bonner A, Hudson M et al. Association of gastroesophageal factors and worsening of forced vital capacity in systemic sclerosis.J Rheumatol. (2013) 40(6):850-858. doi:10.3899/jrheum.120705. Epub 2013 Apr 1. PMID: 23547215
2. Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972-2002.Ann Rheum Dis. (2007) 66(7):940-944. Epub 2007 Feb 28. PMID: 17329309
3. Muangchan C; Canadian Scleroderma Research Group, Baron M, Pope J. The 15% Rule in Scleroderma: The Frequency of Severe Organ Complications in Systemic Sclerosis. A Systematic Review.J Rheumatol. (2013) [Epub ahead of print] PMID: 23858045
4. Pope J. Measures of systemic sclerosis (scleroderma): Health Assessment Questionnaire (HAQ) and Scleroderma HAQ (SHAQ), physician- and patient-rated global assessments, Symptom Burden Index (SBI), University of California, Los Angeles, Scleroderma Clinical Trials Consortium Gastrointestinal Scale (UCLA SCTC GIT) 2.0, Baseline Dyspnea Index (BDI) and Transition Dyspnea Index (TDI) (Mahler's Index), Cambridge Pulmonary Hypertension Outcome Review (CAMPHOR), and Raynaud's Condition Score (RCS).Arthritis Care Res (Hoboken) (2011) 63 Suppl 11:S98-S111. doi: 10.1002/acr.20598.
5. Latsi PI, Wells AU. Evaluation and management of alveolitis and interstitial lung disease in scleroderma.Curr Opin Rheumatol (2003) 15:748-755.
6. Walker KM, Pope J; participating members of the Scleroderma Clinical Trials Consortium (SCTC); Canadian Scleroderma Research Group (CSRG). Treatment of systemic sclerosis complications: what to use when first-line treatment fails--a consensus of systemic sclerosis experts.Semin Arthritis Rheum. (2012) 42(1):42-55. doi: 10.1016/j.semarthrit.2012.01.003. Epub 2012 Mar 29. PMID: 22464314.
7. Tashkin DP, Elashoff R, Clements PJ, et alCyclophosphamide versus placebo in scleroderma lung disease.N Engl J Med. (2006) 354(25):2655-2666. PMID: 16790698
8. Hoyles RK, Ellis RW, Wellsbury J, et alA multicenter, prospective, randomized, double-blind, placebo-controlled trial of corticosteroids and intravenous cyclophosphamide followed by oral azathioprine for the treatment of pulmonary fibrosis in scleroderma.Arthritis Rheum. (2006) 54(12:3962-3970. PMID: 17133610
9. Broad K, Pope JE. The efficacy of treatment for systemic sclerosis interstitial lung disease: results from a meta-analysis.Med Sci Monit. (2010) 16(9)RA187-90. PMID: 20802426.
10. Tashkin DP, Elashoff R, Clements PJ, et al. Effects of 1-year treatment with cyclophosphamide on outcomes at 2 years in scleroderma lung disease.Am J Respir Crit Care Med. (2007) 176(10:1026-1034. Epub 2007 Aug 23. PMID: 17717203
11. Daoussis D, Liossis SN, Tsamandas AC, et alExperience with rituximab in scleroderma: results from a 1-year, proof-of-principle study.Rheumatology (Oxford). (2010) 49(2:271-280. doi: 10.1093/rheumatology/kep093. Epub 2009 May 15.
12. Daoussis D, Liossis SN, Tsamandas AC, et alEffect of long-term treatment with rituximab on pulmonary function and skin fibrosis in patients with diffuse systemic sclerosis.Clin Exp Rheumatol. (2012) 30(2 Suppl 71:S17-22. Epub 2012 May 29.
13. Kowal-Bielecka O, Landewé R, Avouac J, et alEULAR recommendations for the treatment of systemic sclerosis: a report from the EULAR Scleroderma Trials and Research group (EUSTAR).Ann Rheum Dis. (2009) 68(5:620-628. doi: 10.1136/ard.2008.096677. Epub 2009 Jan 15. PMID: 19147617
14. Nadashkevich O, Davis P, Fritzler M, Kovalenko W. A randomized unblinded trial of cyclophosphamide versus azathioprine in the treatment of systemic sclerosis.Clin Rheumatol. (2006) 25(2:205-212. Epub 2005 Oct 14. PMID: 16228107
15. Richeldi L, du Bois RM. Pirfenidone in idiopathic pulmonary fibrosis: the CAPACITY program.Expert Rev Respir Med. (2011) 5(4:473-81. doi: 10.1586/ers.11.52.
16. Sottile PD, Iturbe D, Katsumoto TR, et alOutcomes in systemic sclerosis-related lung disease after lung transplantation.Transplantation. (2013) 95(7:975-980. doi: 10.1097/TP.0b013e3182845f23. PMID: 23545509
17. Christmann RB, Wells AU, Capelozzi VL, Silver RM. Gastroesophageal reflux incites interstitial lung disease in systemic sclerosis: clinical, radiologic, histopathologic, and treatment evidence.Semin Arthritis Rheum. (2010) 40(3:241-249. doi:10.1016/j.semarthrit.2010.03.002. Epub 2010 May 21.
18. Marie I, Dominique S, Levesque H, et alEsophageal involvement and pulmonary manifestations in systemic sclerosis.Arthritis Rheum. (2001) 45(4:346-354
19. Steen VD, Lucas M, Fertig N, Medsger TA Jr. Pulmonary arterial hypertension and severe pulmonary fibrosis in systemic sclerosis patients with a nucleolar antibody.J Rheumatol. (2007) 34(11):2230-2235. Epub 2007 Oct 15. PMID: 17937469
20. Khimdas S, Harding S, Bonner A, et al. Associations with digital ulcers in a large cohort of systemic sclerosis: results from the Canadian Scleroderma Research Group registry.Arthritis Care Res (Hoboken). (2011) 63(1:142-159. doi:10.1002/acr.20336. PMID: 20740608.














