Persistent Headache in a Patient With RA
A 45-year-old Hispanic male physician, a nonsmoker, presented with crippling, nonerosive, seropositive, symmetrical polyarticular arthritis that involved his wrists, hands, elbows, knees, and feet.


A 45-year-old Hispanic male physician, a nonsmoker, presented with crippling, nonerosive, seropositive, symmetrical polyarticular arthritis that involved his wrists, hands, elbows, knees, and feet. Findings included an erythrocyte sedimentation rate (ESR) of 114 mm/h (normal, lower than 23 mm/h) and a weakly positive anti–cyclic citrullinated peptide antibody result. The patient had a history of migraine and tinnitus. He had a previous diagnosis of serous otitis media with middle ear and mastoid effusions on CT scanning; he responded initially to antibiotic and corticosteroid therapy.
A diagnosis of rheumatoid arthritis (RA) was made, and the patient was treated with oral methotrexate and self-injected etanercept. His inflammatory arthritis with disabling musculoskeletal symptoms resolved. However, in spite of a rapid taper of low-dose prednisone early in the disease course, the patient continued to self-prescribe various doses of corticosteroids for worsening headache and tinnitus; all symptoms were relieved.
The patient returned to the clinic and reported recurring eye pain; blurred vision; right-sided and pulsatile facial pain in the V2 and V3 distributions; sinusitis-like pressure; and serous ear drainage with hearing loss, tinnitus, and vertigo. The physical examination revealed sinus tenderness with serous drainage, no lymphadenopathy, normal lung sounds, no scalp tenderness or synovitis, and normal strength. Nailfold capillaroscopy showed irregularly tortuous capillaries but without dropout or dilated loops. The patient’s cerebrospinal fluid (CSF) demonstrated lymphocytosis and normal glucose and protein levels. Chest radiography and urine analysis results were normal. Serum antineutrophil cytoplasmic antibody (c-ANCA) measurement revealed an anti-proteinase 3 (anti-PR3) antibodies level of 86 U/mL and negative results for antimyeloperoxidase antibodies.
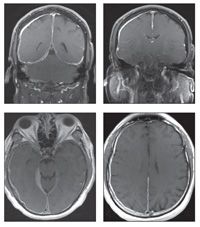
Gadolinium-enhanced coronal (top, left and right) and axial (bottom, left and right) T1-weighted MRI scans of the patient’s head were obtained.
What is your diagnosis?
(Find the answer on the next page.)
The gadolinium-enhanced coronal and axial T1-weighted images show the extent of diffuse dural thickening along the falx cerebri around the right lobe and extending through the tentorium cerebelli bilaterally. These findings along with the patient’s clinical manifestations are consistent with hypertrophic pachymeningitis.


A rare manifestation associated with several rheumatologic diseases, pachymeningitis is characterized by chronic, inflammatory thickening of the dura. The pia and arachnoid also are involved, creating a thick membrane held together with fibrous adhesions.1
The clinical features of pachymeningitis include headache, vision loss, diplopia, papilledema, cranial nerve involvement, ataxia, and seizures. The ESR may be elevated, with elevated protein levels and lymphocytosis in the CSF.2
The differential diagnosis for pachymeningitis is broad, including hematological disease, infection, migraine, malignancy, and iatrogenic drug-induced disease. Given the patient’s serological test results in the setting of an acute inflammatory symmetrical polyarticular arthritis and pachymeningitis, the 2 leading diagnoses were RA and Wegener granulomatosis (WG).
Twenty cases of rheumatoid pachymeningitis have been reported, and they typically have been associated with long-standing disease; seropositivity; erosive, extra-articular features; and rheumatoid nodules in the dura.3 In all cases, the rheumatoid factor and the ESR were elevated. The typical presentation included cranial nerve involvement or headache. Rheumatoid nodules of the dura were found in all autopsy diagnoses, but only rarely with dural biopsy; if present, they were visible on MRI.
Pachymeningitis is found in up to 7% of patients who have WG.4 Typically, there is a male predominance, with limited involvement and only 6 months of active disease before diagnosis. The clinical presentation is chronic headache on nonfocal physical examination. Both positive MRI findings and positive dural biopsy results are 100% sensitive for diagnosis. WG and RA may coexist, although that situation was described in only 6 cases from 1966 to 2003.5
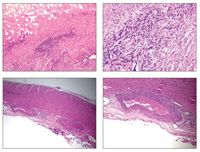
Biopsy of our patient’s dura demonstrated perivascular lymphoplasmacytic infiltration and granulomatous inflammation accompanied by leukocytoclastic debris and necrosis (photomicrographs, hematoxylin and eosin stain) that were most consistent with a small-vessel vasculitis. The only primary systemic vasculitis with an extravascular inflammatory component is WG. Recent data show that within the granulomatous lesions of WG, there is evidence of lymphatic follicles that may lead to elevated levels of inflammatory markers, such as anti-PR3.6 Given the biopsy results and the patient’s presentation, duration of disease in the setting of chronic headache responsive to corticosteroids, and elevated levels of anti-PR3 antibodies, our final diagnosis was WG with pachymeningitis.
Our patient received induction therapy that included a 3-month course of oral cyclophosphamide with high-dose corticosteroids that were tapered slowly. Daily single-strength trimethoprim/sulfamethoxazole also was provided.
With this regimen, the patient reported resolution of his symptoms, including headache; his ESR was 17 mm/h and his c-ANCA level was lower than 6 U/mL. The patient has remained stable on a maintenance regimen of methotrexate, 25 mg/wk, injected subcutaneously.
This case was presented by Jefferson Roberts, MD, an attending rheumatologist at Walter Reed Army Medical Center in Washington, DC, and Alana Wade, a third-year medical student at the Uniformed Services University of Health Sciences in Bethesda, Maryland.
References:
1. Ropper AH, Samuels MA. Disturbances of cerebrospinal fluid and its circulation, including hydrocephalus, pseudotumor cerebri, and low-pressure syndromes. In: Ropper AH, Samuels MA. Adams and Victor’s Principles of Neurology. 9th ed. New York: McGraw-Hill; 2009:591-611.
2. Kupersmith MJ, Martin V, Heller G, et al. Idiopathic hypertrophic pachymeningitis. Neurology. 2004;62:686-694.
3. Thompson MA, Mayer DF. Rheumatoid pachymeningitis presenting with left-sided weakness. Mayo Clin Proc. 2005;80:500.
4. Di Comite G, Bozzolo EP, Praderio L, et al. Meningeal involvement in Wegener’s granulomatosis is associated with localized disease. Clin Exp Rheumatol. 2006;24(2 suppl 41):S60-S64.
5. Douglas G, Bird K, Flume P, et al. Wegener’s granulomatosis in patients with rheumatoid arthritis. J Rheumatol. 2003;30:2064-2069.
6. Mueller A, Holl-Ulrich K, Lamprecht P, Gross WL. Germinal centre-like structures in Wegener’s granuloma: the morphological basis for autoimmunity? Rheumatology (Oxford). 2008;47:1111-1113.














