Pulmonary Hypertension in a Patient With Adult-Onset Still Disease
Adult-onset Still disease (AOSD) is an inflammatory condition characterized by a sudden onset of spiking fever, arthralgia, a salmon-colored evanescent rash, leukocytosis, and an elevated erythrocyte sedimentation rate (ESR) and serum ferritin level.
ABSTRACT: Pulmonary involvement in adult-onset Still disease (AOSD) may take the form of pneumonitis, pleural effusion, and transient pulmonary infiltrates. Pulmonary hypertension (PH) has been reported as a manifestation in only a few cases. We describe the case of a 26-year-old woman with AOSD who was admitted to the hospital with a 1-month history of fever, arthralgia, and excessive fatigue. A reddish, macular, nonblanchable, nonpruritic rash was seen on her extremities. Progressive shortness of breath developed, and the patient received a diagnosis of PH. In the absence of any other cause that could have resulted in PH, we concluded that the patient's PH probably was a result of her AOSD. (J Musculoskel Med. 2011;28:388-390)
______________________________________________________________________________________________
Adult-onset Still disease (AOSD) is an inflammatory condition characterized by a sudden onset of spiking fever, arthralgia, a salmon-colored evanescent rash, leukocytosis, and an elevated erythrocyte sedimentation rate (ESR) and serum ferritin level. Pulmonary involvement may take the form of pneumonitis, pleural effusion, and transient pulmonary infiltrates. Pulmonary hypertension (PH) has been reported as a manifestation of AOSD in only a few cases.
In this article, we describe the case of a 26-year-old woman with AOSD in whom progressive shortness of breath developed and who received a diagnosis of PH. In the absence of any other cause that could have resulted in PH, we concluded that the patient's PH probably was a result of her AOSD.
Case report
A 26-year-old woman of Puerto Rican descent was admitted to the hospital with a 1-month history of fever, arthralgia, and excessive fatigue. A macular rash subsequently developed on her extremities. She had no history of Raynaud phenomenon, malar rash, alopecia, or oral ulcers.
On examination, the patient was tachycardic, with a heart rate of 112 beats per minute (normal, 60 to 100 beats per minute); febrile, with a temperature of 39.3°C (102.8°F; normal, 36.8°C ± 0.7°C or 98.2°F ± 1.3°F); and tachypneic, with a respiratory rate of 18 breaths per minute (normal, 12 to 16 breaths per minute). No lymphadenopathy or hepatosplenomegaly was noted. A reddish, macular, nonblanchable, nonpruritic rash was seen on her extremities. There was no sclerodactyly or skin thickening.
A musculoskeletal examination revealed synovial effusion in both of the patient's wrists, with decreased range of motion. Chest auscultation revealed decreased breath sounds at both lung bases.
The patient's blood work showed leukocytosis, with a leukocyte count of 16.3 × 103/µL (normal, 4.8 to 10.8 × 103/µL), with 87% neutrophils; a normocytic normochromic anemia (hemoglobin level, 9.5 g/dL; normal, 12.3 to 15.3 g/dL); and an elevated ESR (70 mm/h; normal, lower than 21 mm/h) as determined by the Westergren method. Tests for rheumatoid factor and antinuclear antibodies were negative. Anti-ribonucleoprotein, anticentromere, and anti–Scl-70 antibodies were undetectable. The patient's lactate dehydrogenase level was 313 U/L (normal, 100 to 190 U/L), but direct and indirect Coombs test results were negative and the haptoglobin level was within normal limits. The serum ferritin level was 34,100 ng/mL (normal, 10 to 150 ng/mL).
Chest x-ray films revealed bilateral small pleural effusions but no underlying lung parenchymal disease. A biopsy of the patient's rash disclosed superficial and deep perivascular infiltration with neutrophils, findings consistent with AOSD. Treatment with prednisone, 0.5 mg/kg/d, and naproxen was started, and the patient was discharged.
On the patient's follow-up visit, she presented with a recurrence of her rash; joint pain with swelling; and fever spikes, with temperatures as high as 38.9°C (102°F). On examination, she was found to have diffuse cervical lymphadenopathy and splenomegaly.
The patient subsequently was admitted to the hospital. As a part of her workup, a CT scan was obtained; it revealed mildly enlarged mediastinal and hilar lymph nodes and splenomegaly. Lymphoma was suspected, and the patient underwent an excisional biopsy of a cervical lymph node. The biopsy revealed florid atypical hyperplasia, with vascular proliferation and increased atypical immunoblasts, features that have been described in association with Still disease.
The prednisone dosage was increased to 1 mg/kg/d, resulting in resolution of the patient's symptoms. She was monitored as an outpatient, and a prednisone taper was begun.
FIGURE


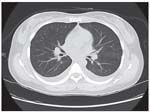
A chest CT scan obtained for our patient with adult-onset Still disease ruled out pulmonary embolism and interstitial lung disease as the cause of her dyspnea.
Six months after the diagnosis, shortness of breath started to develop; it initially was present with exertion but gradually progressed to occur with minimal exertion. A chest x-ray film did not reveal any abnormalities. A CT scan of the patient's chest ruled out pulmonary embolism and interstitial lung disease as the cause of her dyspnea (Figure). Pulmonary function test results were normal.
An echocardiogram revealed normal ejection fraction. There was no evidence of diastolic dysfunction, but right atrial and right ventricular dilatation was noted. Pulmonary artery systolic pressure was 70 mm Hg (normal, 15 to 35 mm Hg), suggesting severe PH. A right heart catheterization revealed main pulmonary artery pressure of 57/28 mm Hg; the mean was 33 mm Hg (normal, less than 25 mm Hg). Pulmonary capillary wedge pressure was 16 mm Hg (normal, 6 to 12 mm Hg). The patient's pulmonary circulation pressures did not improve with nitric oxide.
Discussion
Still disease was first described by George Still in children in 1896.1,2 It was not until 1971 that a similar clinical presentation was first described in young adults and named AOSD.1 There is a bimodal age distribution in AOSD, with one peak between 15 and 25 years and a second peak between 36 and 46 years; the sexes are affected equally.
The development of PH in patients with Still disease has been discussed in only a few case reports,3-5 the first being a case in Japan described by Zen and associates5 in 1990. Subsequently, 2 cases of PH in patients with AOSD were reported in a Taiwan study.6
Although the cause of Still disease is unknown, T-helper cytokines have been reported in the blood of patients,7 and elevated levels of interleukin (IL)-2, IL-6, and IL-18 and tumor necrosis factor α (TNF-α) have been described.7-9 We postulate that these cytokines, TNF-α, and activated B and T cells could be responsible for the pathogenesis of PH.10-12
Primary PH occurs frequently in patients who have scleroderma, especially the limited form of the disease, as well as in those who have mixed connective-tissue disease or systemic lupus erythematosus. There is some evidence in these conditions that the syndrome is mediated by endothelin released by activated or damaged endothelial cells. Of note, a prominent component of our patient's AOSD was a rash that consisted of a perivascular infiltrate, suggesting widespread endothelial damage.
Conclusion
We think that PH is a complication of AOSD as was seen in our patient. Larger studies are required to confirm this finding. In the meantime, we recommend obtaining echocardiograms for patients with AOSD who complain of dyspnea on exertion or a decrease in exercise tolerance.
Problems with/comments about this article? Please send feedback.
References:
References
1. Bywaters EG. Still's disease in the adult. Ann Rheum Dis. 1971;30:121-133.
2. Still GF. On a form of chronic joint disease in children. Med Chir Trans. 1897;80:47-60. (Reprinted in: Arch Dis Child. 1941;16:156-165.)
3. Thakare M, Habibi S, Agrawal S, Narsimulu G. Pulmonary arterial hypertension complicating adult-onset Still's disease. Clin Rheumatol. 2009 Aug 11; [Epub ahead of print].
4. Mubashir E, Ahmed MM, Hayat S, et al. Pulmonary hypertension in a patient with adult-onset Stills disease. Clin Rheumatol. 2007;26:1359-1361.
5. Zen A, Yamashita N, Ueda M, et al. A case of adult Still's disease with pulmonary hypertension [in Japanese]. Ryumachi. 1990;30:45-52.
6. Chen CH, Chen HA, Wang HP, et al. Pulmonary arterial hypertension in autoimmune diseases: an analysis of 19 cases from a medical centre in northern Taiwan. J Microbiol Immunol Infect. 2006;39:162-168.
7. Chen DY, Lan JL, Lin FJ, et al. Predominance of Th1 cytokine in peripheral blood and pathological tissues of patients with active untreated adult onset Still's disease. Ann Rheum Dis. 2004;63:1300-1306.
8. Hoshino T, Ohta A, Yang D, et al. Elevated serum interleukin 6, interferon-gamma and tumor necrosis factor-alpha levels in patients with adult Still's disease. J Rheumatol. 1998;25:396-398.
9. Kawashima M, Yamamura M, Taniai M, et al. Levels of interleukin-18 and its binding inhibitors in the blood circulation of patients with adult-onset Still's disease. Arthritis Rheum. 2001;44:550-560.
10. Budhiraja R, Tuder RM, Hassoun PM. Endothelial dysfunction in pulmonary hypertension. Circulation. 2004;109:159-165.
11. Hassoun PM, Mouthon L, Barberà JA, et al. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol. 2009;54(1 suppl):S10-S19.
12. Dorfmüller P, Perros F, Balabanian K, Humbert M. Inflammation in pulmonary arterial hypertension. Eur Respir J. 2003;22:358-363.















