Skin hardening, but is it systemic sclerosis?
Systemic sclerosis (SSc), or scleroderma, is a condition of connective tissue characterized by inflammation and fibrosis of the skin and internal organs. The disease is rare but associated with significant morbidity and mortality.
Systemic sclerosis (SSc), or scleroderma, is a condition of connective tissue characterized by inflammation and fibrosis of the skin and internal organs. The disease is rare but associated with significant morbidity and mortality.
Early and proper diagnosis of SSc provides the opportunity for meaningful intervention, but several conditions that present with skin hardening may delay and confuse the diagnosis. The more common scleroderma mimics include localized scleroderma (LS), or morphea; nephrogenic fibrosing dermopathy (NFD); eosinophilic fasciitis (EF); chronic graft versus host disease (GVHD); diabetic cheiroarthropathy; and scleredema adultorum. Recognizing a few fundamental facts about SSc helps avoid misdiagnosis. In this article, we discuss the important clinical features of SSc and how this information may help distinguish it from the conditions that have skin induration as a major clinical feature.
EPIDEMIOLOGY
As a group, scleroderma mimics occur as frequently as SSc, if not more frequently. SSc occurs mostly in adults and rarely in children; the incidence is about 19.3 adult cases per million persons per year, and the prevalence is about 276 adult cases per million persons.1 Pea


k incidence occurs in the fifth and sixth decades of life. There is a predominance in women (female to male ratio, 3-8:1).
Although SSc is seen in all racial groups, disease expression differs in various ethnic groups. SSc can be divided into 2 major clinical variants: diffuse and limited (Table 1). Patients of African ancestry are 1.86 times more likely than white patients to have diffuse disease.1
CLINICAL FEATURES
The difference between the diffuse and limited variants of SSc lies in the extent of skin involvement. Diffuse cutaneous SSc involves the extremities (proximal and distal) and the trunk; limited SSc involves the extremities distal to the elbows and knees, with truncal sparing. Both variants affect the face and neck. Patients with CREST (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, telangiectasia) syndrome fall within the limited variant of SSc.2 Sine scleroderma and the overlap syndrome are less common variants of SSc.
Principal features
The most salient features of SSc are skin thickening and the presence of Raynaud phenomenon. Skin change occurs in 3 sequential stages: edematous (characterized by tight, puffy fingers), indurative (characterized by tight, thickened skin that adheres to the underlying subcutis), and atrophic (characterized by softening of the thickened skin; occurs after several years).3 Skin involvement may occur rapidly in the first few months of disease, moving proximally, and may continue for 1 to 3 years, after which it enters the atrophic phase.
Raynaud phenomenon is the result of vasospasm of digital arteries, arterioles, and arteriovenous shunts of the skin; the condition is incited by cold temperatures and emotional stress. Blanching of the digits is followed by cyanosis and, finally, hyperemia. Raynaud phenomenon occurs in 90% to 98% of patients with SSc and may predate the development of skin involvement by several years.4 Severe disease is associated with digital tip reabsorption and ulcerations.
Organ involvement
GI tract involvement is common in SSc, affecting 75% to 90% of patients.5 Smooth muscle atrophy and submucosal fibrosis result in dysmotility of the entire GI tract. Esophageal dysfunction results in dysphagia and dyspepsia; gastric involvement produces delayed emptying, with nausea, bloating, and early satiety. Small-intestine dysmotility results in abdominal pain, diarrhea, and pain caused by malabsorption due to bacterial overgrowth and pseudo-obstruction. Colonic involvement may manifest as constipation and megacolon.
Pulmonary abnormalities, particularly restrictive lung disease, are common manifestations in diffuse SSc, occurring in 90% of patients.6 Moderate to severe interstitial lung disease develops in about 40% of patients.6 The most common symptoms are shortness of breath and, later, a nonproductive cough. Pulmonary artery hypertension (PAH) is more common in patients with limited SSc; it usually presents about 10 years after the original diagnosis. PAH also occurs in up to 10% to 15% of patients with diffuse cutaneous SSc, usually in the setting of pulmonary fibrosis.7 Pulmonary involvement is the leading cause of death in patients with SSc. PAH accounts for about 30% of deaths; in patients with limited SSc, the median survival time is only 12 months.8
Other important organ involvement includes the heart and kidneys. Myocardial fibrosis may be seen with cardiac arrhythmias, pericarditis, and ventricular heart failure; these are poor prognostic indicators.9 Renal failure with hypertension, or scleroderma renal crisis, is seen most frequently in the diffuse variant of SSc. Microangiopathic hemolytic anemia with thrombocytopenia often occurs concomitantly. Quick recognition of new-onset hypertension or deteriorating renal function or both can lead to effective intervention with angiotensin-converting enzyme inhibitors.10
Histopathology
SSc is associated with vascular intimal proliferation, with luminal narrowing, mononuclear cellular infiltrates, and increased collagen deposition by fibroblasts. These findings are not specific and cannot be relied on to distinguish SSc from other fibrosing conditions of the skin. The immune system plays an important role in the pathogenesis of SSc, and there is evidence of both humoral and cellular immune involvement.
Antinuclear antibodies (ANAs) occur in 70% to 95% of patients with SSc.11 The mechanism by which autoantibodies induce damage is not clear. Specific autoantibodies help distinguish variants of SSc. Anti-topoisomerase I, or Scl-70, is present in 30% to 40% of diffuse SSc disease cases, and the anticentromere pattern is seen in 70% to 90% of the limited variant cases.12 Activated T cells (CD4+ and CD8+) dominate the inflammatory process in the tissues of patients with SSc.13 SSc is almost always associated with Raynaud phenomenon and characteristic skin changes in the hands and face. Arthralgia, myalgia, and esophageal symptoms are common. Injuries to the lungs, heart, and kidneys are more frequent in the diffuse variant. These characteristics help segregate conditions that at first glance can be confused with SSc.
MIMICKING CONDITIONS
Several conditions may present with significant skin induration. They include other immune-mediated conditions (chronic GVHD, EF, LS), deposition disorders (diabetes mellitus [DM]), and exposure to external agents (nephrogenic systemic fibrosis [NSF]) (Table 2).
Localized scleroderma
LS represents a group of distinct conditions in which skin and subcutaneous tissue involvement is predominant and internal organ involvement is uncommon. LS is considered clinically distinct from SSc. However, autoantibodies are present in LS, which suggests that the separation from SSc is not so clear-cut. These diseases may represent the extremes of a single disease spectrum.
The pathophysiology of LS involves abnormal collagen deposition. Although the etiology is unknown, data suggest an autoimmune process. Most forms of LS involve the dermis, but other forms involve the deeper tissues, such as the fascia and bone, resulting in deformities.
LS is more common than SSc; the incidence is about 3 cases per 100,000 persons.14 Like SSc, LS is predominant in women and occurs in all age-groups (mean age at onset, 20 to 40 years in adults and 7 years in children).
Various classification systems have attempted to categorize the subtypes of LS. The most widely used classification system divides LS into 5 broad categories: plaque, generalized, bullous, linear, and deep morphea.15 A newer, more comprehensive scheme for classification was proposed recently.16 Plaque morphea is the most common type in adult patients, accounting for two-thirds of cases. Generalized morphea is the second most common form and the easiest to confuse with SSc.
Plaque morphea. This subtype is superficial and usually confined to the dermis, although occasionally it involves the superficial panniculus. The lesions usually begin as an area of erythema or induration with a violaceous border, often more than 1 cm in diameter, that involves 1 or 2 anatomical sites. The lesions may soften over time and turn yellow, eventually becoming sclerotic with loss of adnexal structures (sweat glands and hair) and with associated pigment changes.14 Plaque morphea usually is self-limited (average duration, 3 to 5 years). In rare cases, it progresses to SSc.
Generalized morphea. This is the designation when 2 or more areas of induration larger than 3 cm coalesce, forming confluent lesions at 2 or more anatomical locations. They include the head and neck, left and right upper extremities, left and right lower extremities, and anterior and posterior trunk (Figure 1).


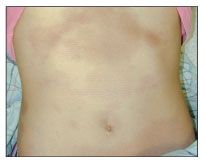
Figure 1 –In patients with generalized morphea, large confluent areas of induration involve 2 or more anatomical sites. In this case, the anterior abdomen and both lower extremities (thighs) are affected.
Bullous morphea. This subtype involves formation of tense subepidermal bullae within plaques of morphea. The cause of bullae is not known, but they have been thought to result from local trauma or lymphatic obstruction caused by scleroderma.15
Linear morphea. This is the most common subtype in children, accounting for about one-third of cases.14 The lesions involve the dermis, subcutaneous tissues, muscle, and even bone. Linear morphea usually affects the extremities and is unilateral in 95% of cases.14 The lesions do not follow a dermatomal distribution. The face and scalp can be involved, with atrophy and indentation of the underlying bone reminiscent of a sword strike injury (en coup de sabre). Facial involvement is associated with ocular deformities, such as ptosis and eyebrow/lash loss, and visual disturbances; cognitive abnormalities; and seizures. Morbidity results from limb deformities, joint contractures, and limb atrophy.
Deep morphea. In contrast to plaque morphea, which primarily affects the dermis, deep morphea involves the deep dermis, subcutaneous tissue, fascia, or superficial muscle. The lesions of deep morphea are more diffuse, which differentiates it from linear morphea.
The diagnosis of LS is based on the clinical presentation and, occasionally, skin biopsy. The epidermis is normal. The dermis may have perivascular lymphocytic infiltrates, as well as increased and thickened collagen bundles, with entrapment of sweat glands.
Clinically, LS differs from SSc with the absence of Raynaud phenomenon, the sparing of hands and feet, and the rare and atypical involvement of internal organs. Although morbidity can be significant in some cases (linear scleroderma), the prognosis is generally good. Many treatment modalities have been tried unsuccessfully, but there has been some success with topical corticosteroids, UV-A, and pulse corticosteroids with methotrexate.17
Nephrogenic systemic fibrosis
Fibrotic disorders have been seen with medications (eg, bleomycin) and also unknown contaminants found in preparations of tryptophan (eosinophilia-myalgia syndrome) and rapeseed oil (toxic oil syndrome). The latest disorder associated with an external agent, NSF, was identified in 2000; it is named nephrogenic systemic fibrosis because it has been seen in patients with chronic renal failure.
The etiology of NSF is unknown, but it is associated with exposure to gadolinium-based contrast agents in patients who have acute or chronic renal insufficiency (glomerular filtration rate [GFR], lower than 30 mL/min/1.73 m2).17,18 The time between gadolinium exposure to the development of NSF is variable; it has been noted to occur 2 to 75 days after exposure. NSF may occur in children but is seen more frequently in adults. It occurs with an equal sex distribution and in persons with various ethnic backgrounds.
NSF is characterized by the onset of painful or pruritic edema of the extremities. The lesions may have an erythematous to brawny hyperpigmentation; over time, the lesions take on a woody appearance (Figure 2).


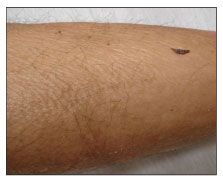
Figure 2 – Skin induration of the forearm with lumpy nodular plaques may be present in nephrogenic systemic fibrosis. Pigmentary changes and flexion contractures of the extremities may occur. (Photograph courtesy of Dr Michael Kraus.)
Various presentations have been reported, ranging from subtle, superficial papules and plaques to deep dermal induration and contractures. Lesions usually involve the extremities (66% to 85% of cases) and, less often, the hands, feet, and trunk (23% to 34% of cases).19 The face and buttocks are affected infrequently (3% to 9% of cases). The skin changes may progress at a variable rate over the course of days to weeks. Lesions may stabilize, but complete resolution is rare. NSF does not progress to systemic involvement, although cases have been reported with subclinical involvement of the heart, lungs, esophageal muscle, skeletal muscle, and diaphragm.19
There are no specific laboratory abnormalities with NSF, although the erythrocyte sedimentation rate (ESR) and C-reactive protein level usually are elevated.18 Autoantibodies are not found. Histological findings vary with the age of the lesion. Early lesions demonstrate narrow collagen bundles, with marked edema and mucin separating the bundles; older lesions contain thickened collagen bundles. Neutrophils and eosinophils are not present in NSF but, rarely, plasma cells are noted.19
NSF differs from SSc with the general absence of Raynaud phenomenon, systemic involvement, and autoantibodies. NSF differs from SSc histologically with its significant cellularity and general absence of plasma cells, neutrophils, and eosinophils.
There is no treatment for patients with NSF. The goal is to reduce the risk of NSF. Gadolinium-based contrast media should be avoided in patients with acute or chronic renal insufficiency (GFR, lower than 30 mL/min/1.73 m2), or renal insufficiency resulting from hepatorenal syndrome, and in the perioperative liver transplantation period. Other factors that may play a role include high-dose epoetin therapy, high calcium and phosphate levels, metabolic acidosis, and recent surgical or thrombotic events.20,21
Immediate hemodialysis within 2 to 4 hours after exposure, followed by additional sessions over the next 2 to 4 days, is recommended. However, whether hemodialysis actually prevents the development of NSF is not known.
Chronic graft versus host disease
This is a common complication after an allogeneic hematopoietic stem cell transplantation (HSCT), occurring in 60% to 80% of long-term survivors.22 Chronic GVHD is manifested primarily in the skin, oral mucosa, GI tract, and liver. The incidence of cutaneous chronic GVHD has increased because there has been a shift to using reduced-intensity preparative regimens. More patients are therefore eligible for HSCT, and more patients survive long enough for chronic GVHD to develop.22 Risk factors for chronic GVHD include previous acute GVHD, human leukocyte antigen disparity, older age of patient or donor, and the use of non-T-cell–depleted marrow.23,24
GVHD has been classified as acute GVHD, occurring within the first 100 days after an allogeneic HSCT, or chronic GVHD, occurring after the first 100 days post-transplant. Cutaneous chronic GVHD typically is divided into 2 categories, lichenoid and sclerodermoid. Although sclerodermoid GVHD often is preceded by a lichenoid phase, the sclerodermoid and lichenoid lesions may occur independently.22 Lichenoid GVHD often occurs early in the course of chronic cutaneous GVHD. The lesions are pink to violaceous, scaly papules that occur in the periorbital area and on the ears, palms, and soles.25
Sclerodermoid GVHD develops in about 3% of patients with GVHD.26 This condition may be localized or generalized (defined as more than 2 anatomical sites); it usually presents as inflammatory plaques similar to those seen in morphea. Over time, the lesions become sclerotic with associated atrophy and pigment changes.24 Joint contractures are a late complication of sclerodermoid GVHD.
Sclerodermoid GVHD differs from SSc both clinically and histologically. Patients with this condition rarely have Raynaud phenomenon or acrosclerosis. Other manifestations (eg, xerophthalmia, cholestasis, esophageal involvement, obliterative bronchiolitis, and myositis) might be confusing were it not for the history of a preceding HSCT.
Sclerodermoid GVHD histologically demonstrates marked epidermal atrophy, destruction of appendageal structures, superficial collagen fibrosis, and linearization of the dermoepidermal junction. Granular IgM deposits have been noted at the dermoepidermal junction in 86% of patients with sclerodermoid GVHD.27
Treatment of patients with sclerodermoid GVHD involves systemic immunosuppression with prednisone and cyclosporine. Phototherapy may play a role in the management of cutaneous sclerodermoid GVHD, but this treatment approach is controversial.26 Newer, more promising therapeutic strategies include thalidomide, anti–tumor necrosis factor-α blockade, and B-cell depletion.
Diabetic cheiroarthropathy and scleredema adultorum
Diabetic cheiroarthropathy is defined by stiffness of the hands and flexion contractures seen in patients with long-standing type 1 DM, as well as with type 2 DM. It has been evident in 9% to 32% of patients with type 1 DM, but under more careful assessment, a generalized reduction of joint mobility was documented in all patients with type 1 DM.28 Diabetic cheiroarthropathy was a significant feature of long-standing disease (more than 9 years).
Diabetic cheiroarthropathy also is characterized by thick, waxy skin associated with sclerosis of tendon sheaths. This may be apparent clinically with the "prayer sign"-the patient cannot press the palms and fingers together completely (Figure 3). The cause of diabetic cheiroarthropathy is thought to be multifactorial; it probably involves increased glycation of collagen, decreased collagen degradation, diabetic microangiopathy, and diabetic neuropathy. There is no specific treatment for patients with diabetic cheiroarthropathy.29



Figure 3 – Diabetic cheiroarthropathy often results in the "prayer sign." Accumulation of collagen in skin and flexor tendons in diabetes mellitus leads to loss of full extension of the fingers.
Scleredema adultorum (of Buschke) is characterized by firm nonpitting edema that typically begins at the neck and spreads to the face, scalp, shoulders and trunk.30 The hands and feet are spared. The condition is seen as a complication of both types 1 and 2 DM, accounting for about half of cases. This form of skin induration also has been described in association in hypergammaglobulinemia with paraproteinemia and after streptococcal infections.31,32
Eosinophilic fasciitis
This rare fibrosing disorder affects the deep fascial layer, leaving the overlying uninvolved skin with a wrinkled, woody appearance. The age of affected persons ranges from 11 to 72 years (mean, 47 years).33 The cause is unknown, but EF has been temporally related to strenuous exercise; the use of medications (phenytoin, antituberculous agents); and infections, particularly Borrelia burgdorferi infection.33
EF is characterized by tender edema of the extremities with a rapid onset (days to weeks); the face, hands, and feet usually are spared. After the initial tenderness and edema, a coarse, "orange peel" appearance develops in the skin and there is subcutaneous induration.
Musculoskeletal organ involvement includes carpal tunnel syndrome (25% of patients), arthritis, myopathy, and contractures.33-35 In most patients, there is no Raynaud phenomenon or internal organ involvement.
Histologically, there is inflammation and sclerosis of the fascia with infiltration of lymphocytes and plasma cells. The epidermis and dermis are spared. Therefore, a full-thickness skin biopsy extending down to the muscle is necessary to make a histological diagnosis.36
Laboratory evaluation usually demonstrates blood eosinophilia, an elevated ESR, and hypergammaglobulinemia. Abnormal serology (eg, ANAs and rheumatoid factor) is not present.
EF can be self-limited, resolving in 3 to 5 years, but many patients require therapy with corticosteroids. A response may be seen within weeks, with a decrease in the accompanying eosinophilia. Resistant cases have been treated with numerous immunosuppressive agents, with mixed success. Hematological disorders, including malignant myelodysplasic and lymphoproliferative disorders, may develop in about 10% of patients.35,37 Morphealike lesions and truncal involvement are associated with greater residual fibrosis.38
EF differs from SSc with the absence of Raynaud phenomenon, the presence of normal nail fold capillaries, a lack of epidermal and dermal involvement, and the general absence of systemic involvement. Eosinophilia is uncommon in SSc.35 ANAs and anti Scl-70 autoantibodies are not found in EF.


SUMMARY
Skin induration is not synonymous with SSc. Numerous conditions possess this feature. Paying careful attention to the accompanying historical and clinical characteristics helps considerably in the differential diagnosis (Table 3). The absence of Raynaud phenomenon, a lack of skin involvement in the hands, the absence of internal organ involvement, and a normal ANA test result would justify hesitation in making a diagnosis of SSc. Early consultation with rheumatologic and dermatological specialists would greatly expedite proper diagnosis and management.
References:
References1. Mayes MD, Lacey JV Jr, Beebe-Dimmer J, et al. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum. 2003;48:2246-2255.
2. LeRoy EC, Black C, Fleischmajer R, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol. 1988;52:202-205.
3. Clements PJ, Medsger TA Jr, Feghali CA. Cutaneous involvement in systemic sclerosis. In: Clements PJ, Furst DE, eds. Systemic Sclerosis. Philadelphia: Lippincott Williams & Wilkins; 2004:129-150.
4. Wigley FM. Clinical practice: Raynaud’s phenomenon. N Engl J Med. 2002;347:1001-1008.
5. Domsic R, Fasanella K Bielefeldt K. Gastrointestinal manifestations of systemic sclerosis. Dig Dis Sci. 2008;53:1163-1174.
6. Ostojic P, Cerinic MM, Silver R, et al. Interstitial lung disease in systemic sclerosis. Lung. 2007;185:211-220.
7. MacGregor AJ, Canavan R, Knight C, et al. Pulmonary hypertension in systemic sclerosis: risk factors for progression and consequences for survival. Rheumatology (Oxford). 2001;40:453-459.
8. Bull TM. Screening and therapy of pulmonary hypertension in systemic sclerosis. Curr Opin Rheumatol. 2007;19:598-603.
9. Janosik DL, Osborn TG, Moore TL, et al. Heart disease in systemic sclerosis. Semin Arthritis Rheum. 1989;19:191-200.
10. Steen VD. Scleroderma renal crisis. Rheum Dis Clin North Am. 2003;29:315-333.
11. Sapadin AN, Esser AC, Fleischmajer R. Immunopathogenesis of scleroderma-evolving concepts. Mt Sinai J Med. 2001;68:233-242.
12. Steen VD. Autoantibodies in systemic sclerosis. Semin Arthritis Rheum. 2005;35:35-42.
13. White B. Immunologic aspects of scleroderma. Curr Opin Rheumatol. 1995;7:541-545.
14. Laxer R, Zulian F. Localized scleroderma. Curr Opin Rheumatol. 2006;18:606-613.
15. Peterson LS, Nelson AM, Su WP. Classification of morphea (localized scleroderma). Mayo Clin Proc. 1995;70:1068-1076.
16. Zulian F. New developments in localized scleroderma. Curr Opin Rheumatol. 2008;20:601-607.
17. Chung L, Lin J, Furst DE, Fiorentino D. Systemic and localized scleroderma. Clin Dermatol. 2006:24:374-392.
18. Marckmann P, Skov L, Rossen K, et al. Nephrogenic systemic fibrosis: suspected causative role of gadodiamide used for contrast-enhanced magnetic resonance imaging. J Am Soc Nephrol. 2006;17:2359-2362.
19. Boyd AS, Zic JA, Abraham JL. Gadolinium deposition in nephrogenic fibrosing dermopathy. J Am Acad Dermatol. 2007;56:27-30.
20. Cowper SE, Rabach M, Girardi M. Clinical and histological findings in nephrogenic systemic fibrosis. Eur J Radiol. 2008;66:191-199.
21. Saab G, Abu-Alfa A. Nephrogenic systemic fibrosis-implications for nephrologists. Eur J Radiol. 2008;66:208-212.
22. Schaffer JV, McNiff JM, Seropian S, et al. Lichen sclerosus and eosinophilic fasciitis as manifestations of chronic graft-versus-host disease: expanding the sclerodermoid spectrum. J Am Acad Dermatol. 2005;53:591-601.
23. Bhushan V, Collins RH Jr. Chronic graft-vs-host disease. JAMA. 2003;290:2599-2603.
24. White JM, Creamer D, du Vivier AW, et al. Sclerodermatous graft-versus-host disease: clinical spectrum and therapeutic challenges. Br J Dermatol. 2007;156:1032-1038.
25. Aractingi S, Chosidow O. Cutaneous graft-versus-host disease. Arch Dermatol. 1998;134:602-612.
26. Ratanatharathorn V, Ayash L, Lazarus HM, et al. Chronic graft-versus-host disease: clinical manifestation and therapy. Bone Marrow Transplant. 2001;28:121-129.
27. Peñas PF, Jones-Caballero M, Aragüés M, et al. Sclerodermatous graft-vs-host disease: clinical and pathological study of 17 patients. Arch Dermatol. 2002;138:924-934.
28. Campbell RR, Hawkins SJ, Maddison PJ, Reckless JP. Limited joint mobility in diabetes mellitus. Ann Rheum Dis. 1985;44:93-97.
29. Beers WH, Ince A, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum. 2006;35:355-359.
30. Lewerenz V, Ruzicka T. Scleredema adultorum associated with type 2 diabetes mellitus: a report of three cases. J Eur Acad Dermatol Venereol. 2007;21:560-561.
31. Meguerditchian C, Jacquet P, Béliard S, et al. Scleredema adultorum of Buschke: an under recognized skin complication of diabetes. Diabetes Metab. 2006;32(5, pt 1):481-484.
32. Kim RP. The musculoskeletal complications of diabetes. Curr Diab Rep. 2002;2:49-52.
33. Maddison PJ. Eosinophilic fasciitis. Br J Rheumatol. 1990;29:81-82.
34. Maddison PJ. Mixed connective tissue disease, overlap syndromes, and eosinophilic fasciitis. Ann Rheum Dis. 1991;50(suppl 4):887-893.
35. Lakhanpal S, Ginsburg WW, Michet CJ, et al. Eosinophilic fasciitis: clinical spectrum and therapeutic response in 52 cases. Semin Arthritis Rheum. 1988;17:221-231.
36. Bennett RM, Herron A, Keogh L. Eosinophilic fasciitis: case report and review of the literature. Ann Rheum Dis. 1977;36:354-359.
37. Khanna D, Verity A, Grossman JM. Eosinophilic fasciitis with multiple myeloma: a new haematological association. Ann Rheum Dis. 2002;61:1111-1112.
38. Endo Y, Tamura A, Matsushima Y, et al. Eosinophilic fasciitis: report of two cases and a systemic review of the literature dealing with clinical variables that predict outcome. Clin Rheumatol. 2007;26:1445-1451.

















