Early Identification of Juvenile Idiopathic Arthritis
A description and guide to manifestations and differential diagnosis of the many subtypes of juvenile idiopathic arthritis (JIA): oligoarticular, polyarticular, systemic, enthesitis-related, and psoriatric JIA.
Chronic arthritis, a complex condition, is the leading cause of autoimmune disease in children, affecting about 1 in 1000 worldwide.1 Juvenile idiopathic arthritis (JIA) collectively describes the group of arthritides of unknown causes that last more than 6 weeks in children younger than 16 years. Consisting of several subtypes, JIA is a significant cause of short- and long-term disability.
The goal of JIA management is to achieve remission. However, with less than full understanding of the disease pathogenesis and a lack of controlled trials, clinicians are left with little evidence to formulate decisions about effective treatment.2 Without appropriate treatment, JIA may result in permanent disability from joint destruction, growth retardation, or blindness from chronic anterior uveitis.1
However, early disease identification and treatment lead to improved quality of life for children with JIA. Recent studies showed that as many as 50% of children who have JIA enter adulthood with active disease,3 and evidence of ongoing disease into adulthood has led to a shift in the treatment paradigm.
In this 2-part article, we discuss early diagnosis and management of JIA. This first part reviews the epidemiology, pathogenesis, clinical manifestations, classification, differential diagnosis, and laboratory analysis of JIA. In the second part, to appear in a later issue of this journal, we will explore the treatment options.
EPIDEMIOLOGY
The reported incidence of JIA varies from 1 to 22 cases per 100,000 children, with a prevalence of 8 to 150 cases per 100,000.4,5 Chronic arthritis in children is defined arbitrarily as arthritis onset earlier than age 16 years. Onset before age 6 months is very unusual. The highest frequency occurs between 1 and 3 years, although age varies with subtype. Twice as many girls as boys are affected with chronic arthritis, but more boys present with spondyloarthropathy.4
CAUSES/PATHOGENESIS
The cause of JIA remains unknown, but it seems to result from a complex genetic trait with an immunoinflammatory pathogenesis, possibly influenced by external antigens.4,5 Abnormalities both cell-mediated (via inflammatory cells, cytokines, and activated T cells) and humoral (via autoantibodies) are implicated. The various phenotypes of the disease point toward interactions of multiple genes affecting immunity and inflammation.
There are HLA associations for each JIA subtype, the strongest in children with oligoarticular disease. HLA-B27 may contribute to the disease pathogenesis via molecular mimicry among patients with enthesitis-related arthritis (ERA).
Immune dysfunction in JIA is evident with the presence of autoantibodies, such as antinuclear antibodies (ANAs)-present in 40% of patients-and rheumatoid factor (RF)-present in 5% to 10% of patients.4,5 Although undoubtedly there are genetic predispositions to JIA, other contributing factors have been suggested, such as environmental agents, infectious vectors, trauma, psychological stress, and hormonal abnormalities.4,5
CLINICAL MANIFESTATIONS
The diagnosis is made using a detailed history and physical examination in concordance with appropriate laboratory and radiographic testing. Common features are morning stiffness and gelling after inactivity. Improvement frequently occurs as the day progresses. Most young children do not complain of stiffness or pain, but there may be changes in gait or function.2
Any joint may be affected, but large joints are involved more frequently. An arthritic joint exhibits inflammation, with swelling, heat, pain, and decreased range of motion. Although the joints often are warm, they are not erythematous.4,6
Erythema and extreme pain may be experienced in septic arthritis and in the reactive arthritides. Attention should be paid to less obvious joints, such as the temporomandibular joint, because it is involved frequently and may include significant micrognathia and difficulty in chewing.4
Other joints that may be affected in JIA include the cervical, thoracic, and lumbosacral spine; hips; and shoulders. Synovitis in these joints cannot be visualized, but there is limited range of motion or guarding on examination. The sternoclavicular, acromioclavicular, and sternomanubrial joints rarely are affected.2,4
In addition to musculoskeletal symptoms, patients may present with anorexia, weight loss, and growth failure. Significant fatigue is more common in patients who have systemic disease or polyarticular JIA. The examiner should evaluate closely for abnormal pupils, rash, lymphadenopathy, hepatomegaly, splenomegaly, and pericardial or pleural rubs.


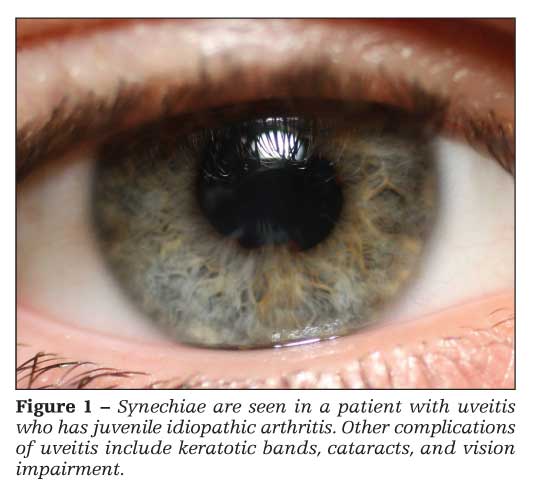
Uveitis
Chronic anterior, nongranulomatous uveitis (iridocyclitis) occurs in about 20% of patients with oligoarticular JIA and 10% of patients with polyarticular JIA (Figure 1).5 Uveitis is most common in girls who have ANA-positive oligoarticular JIA. Patients usually are asymptomatic, but they may exhibit conjunctivitis, pupil asymmetry, and eye pain. Patients who have JIA should be screened routinely. Complications of uveitis include synechiae, keratotic bands, cataracts, and vision impairment.7
CLASSIFICATION
The International League of Associations for Rheumatology classification of JIA includes 7 subtypes: oligoarticular JIA, polyarticular RF-positive and RF-negative JIA, systemic JIA, psoriatic JIA, ERA, and undifferentiated arthritis.8 Oligoarticular JIA occurs most frequently (50% to 60% of cases), followed by polyarticular JIA (30% to 35%), systemic JIA (10% to 20%), psoriatic JIA (2% to 15%), and ERA (1% to 7%).4
Oligoarticular JIA
This subtype is defined as arthritis that affects 4 or fewer joints during the first 6 months of disease. Patients often are young girls, aged 1 to 4 years, with large joint involvement of the lower extremities, such as the knees or ankles; one-half present with arthritis in a single knee. About one-fourth have no pain, and most have little difficulty in functioning. About 70% are ANA-positive, and none are RF-positive. This patient group, especially young girls who have a positive ANA, are at greater risk for asymptomatic uveitis.
Arthritis that remains confined to 4 or fewer joints is classified as persistent oligoarticular JIA. Patients who recruit 5 or more joints after the first 6 months are considered to have extended oligoarticular JIA. Extended disease will develop in up to half of patients; they have only a 12% remission rate, compared with a 75% remission rate in patients with persistent oligoarticular disease.2,4,5
Polyarticular JIA
This subtype is defined as arthritis that affects 5 or more joints within the first 6 months of disease. It is divided into 2 groups, RF-positive and RF-negative. Both affect girls more frequently than boys.
Polyarthritis typically develops in RF-seronegative patients early in childhood, and they have a better prognosis. Seropositive patients typically are adolescent girls with severe symmetrical erosive disease of the small and large joints. The HLA associations in these patients are the same as those in adults with rheumatoid arthritis and probably represent an early presentation of this disease.5 Chronic uveitis is less common in polyarticular than in oligoarticular JIA, but patients still require routine screening.
Systemic JIA
This subtype is characterized by 2 weeks of fever, high quotidian spikes for at least 3 days, and at least 1 of the following: evanescent and erythematous rash, lymphadenopathy, hepatosplenomegaly, and serositis (often pericarditis or pleuritis). Systemic JIA affects boys and girls equally; it may occur at any time but is most common in the early years of childhood. Children appear quite ill when febrile but are well-appearing when they defervesce.
Because arthritis may not occur for several months after the initial symptoms, a definitive diagnosis may be delayed. The arthritis usually is polyarticular. Typical laboratory findings include anemia, leukocytosis, thrombocytosis, elevated liver enzyme levels, and elevated acute phase reactant levels (the C-reactive protein [CRP] level usually is disproportionately higher than the erythrocyte sedimentation rate [ESR]). Results of the ANA titer are rarely positive.
A rapidly improved ESR may not be indicative of disease quiescence. It is associated with macrophage activation syndrome, a life-threatening complication of systemic JIA characterized by hemophagocytosis. Patients are ill, and a severe coagulopathy develops. Sixty percent to 85% of patients with systemic JIA go into remission, but a chronic destructive polyarthritis develops in up to 37% of patients.4-6
Enthesitis-related arthritis
ERA occurs most frequently in boys older than 8 years. This subtype includes juvenile ankylosing spondylitis and arthritis associated with inflammatory bowel disease (IBD). There is a high frequency of HLA-B27 association and a positive family history.
Patients with ERA demonstrate pain, stiffness and, eventually, decreased mobility of the back. Peripheral arthritis typically precedes axial involvement. The hallmark of this form is enthesitis (inflammation at tendon insertions). Sacroiliac arthritis develops in many patients; it can be appreciated on radiographs as joint-space narrowing, erosions, sclerosis, osteoporosis, and fusion (a late finding).5
Articular manifestations may be the first signs of IBD. Therefore, screening for GI symptoms, growth failure, erythema nodosum, and aphthous stomatitis is important.
Acute uveitis develops in about 27% of patients with ERA. However, the uveitis in ERA is symptomatic, with redness, pain, and photophobia, and typically is unilateral and recurrent.2,4,6
Psoriatic JIA
This subtype is arthritis that occurs in conjunction with psoriasis or psoriatic features. It is more common in girls and typically peaks in mid-childhood. Diagnosis may be difficult because the arthritis may precede skin findings by years.


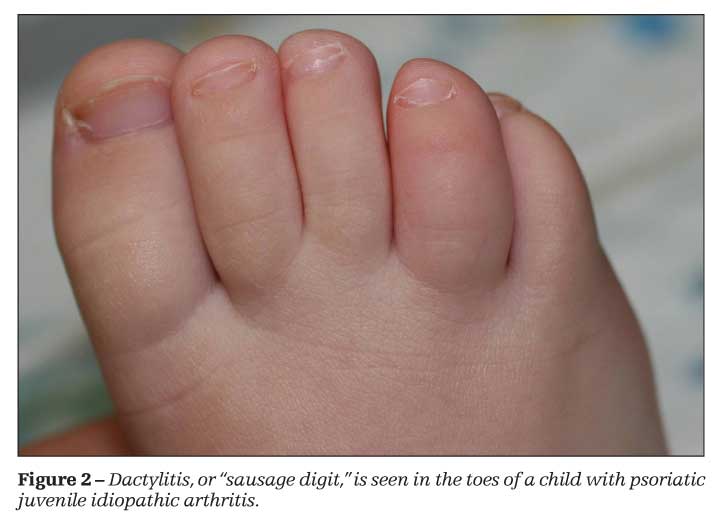
Psoriatic JIA is an asymmetrical arthritis that affects the knees, ankles, and small joints of the hands and feet. Dactylitis, or diffuse swelling of the fingers and toes-often described as a “sausage digit”-is not uncommon (Figure 2). Extra-articular manifestations include nail changes, such as pitting and onycholysis, and asymptomatic uveitis. The RF is negative, but ANAs may be positive.4,5
Undifferentiated arthritis
This subtype of JIA includes patients who do not meet criteria for any category. Or, they meet the criteria for more than 1 category but with either a close relative who has psoriasis or the presence of RF.
DIFFERENTIAL
A diagnosis of JIA is made after other causes of arthritis have been excluded. Clinical findings (eg, rash, systemic illness, arthritis character and duration, length of fever, and evidence of a previous infection) help differentiate JIA from other underlying causes of symptoms.


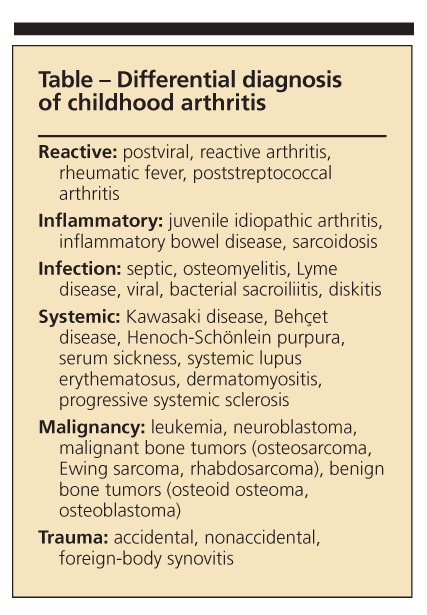
The differential diagnosis of arthritis may be subdivided into general categories of reactive arthritis (usually postenteric infection but may be post–sexually transmitted disease), inflammatory or autoimmune disease (lupus, dermatomyositis, IBD, cystic fibrosis), infection (especially Lyme), systemic illness (sarcoidosis), neoplasia (pigmented villonodular synovitis, leukemia, lymphoma), and trauma (occult fracture, foreign body) (Table).
Often, a distinction may be made between oligoarticular JIA and other causes on the basis of history of antecedent infection and arthritis less than 6 weeks duration. Septic arthritis is considered in a patient with acute onset of fever, joint swelling, and erythema in addition to exquisite joint pain. Synovial fluid should be obtained and cultured and antibiotics should be started immediately because septic arthritis may lead quickly to joint destruction. Patients with gonococcal arthritis may have fever, chills, and rash in addition to arthritis. A complete sexual history is pertinent.5
A misdiagnosis of JIA may be made when children present with arthralgia and bone pain. Bone tenderness should raise suspicion for underlying malignancy. There often is a discrepancy in laboratory testing in these patients. They may have thrombocytopenia relative to an elevated ESR. If there is an elevated ESR, thrombocytosis would be expected because both are acute phase reactants.
Multisystem rheumatologic disease may be distinguished from JIA by clinical features and laboratory findings. Systemic lupus erythematosus (SLE) often presents in adolescence with fever, rash, and nonerosive arthritis. The ANA is positive in SLE but also may be present in oligoarticular and polyarticular JIA. Both SLE and systemic-onset JIA may manifest as fever and serositis, but the skin findings are different. In addition, malar rash, nephritis, cytopenia, hypocomplementemia, and anti–double-stranded DNA and other autoantibodies are seen only in SLE. Patients with dermatomyositis may have polyarthritis early, but development of pathognomonic skin findings and muscle weakness rule out JIA.1,5
Orthopedic complaints associated with patellofemoral syndrome and Osgood-Schlatter disease are common in active adolescents who complain of knee pain. They typically do not awaken with morning stiffness, and their pain worsens with exercise. Musculoskeletal pain without arthritis is seen in chronic pain syndromes, such as reflex neurovascular dystrophy. Features include allodynia (pain to light touch) and pain out of proportion to examination findings.
LABORATORY ANALYSIS
Although directed laboratory tests may support the diagnosis of JIA or provide evidence of inflammation, there are no laboratory tests to confirm the diagnosis. Initial laboratory evaluation for a child with suspected arthritis should include a complete blood cell count with differential, CRP level, and ESR. In addition to documenting the presence or absence of systemic inflammation, these tests can identify hematological abnormalities that may reflect malignancy. Serological testing for Lyme disease is recommended in children who have oligoarthritis and live in areas in which Lyme disease is endemic. Among children with clinically confirmed JIA, an ANA test can help determine the risk of uveitis. The ANA result may be positive in up to 20% of normal children; thus, an elevated ANA level is not diagnostic of JIA, and care should be taken in interpreting a positive result in someone who does not have arthritis.
RF positivity is infrequent in JIA and unusual in children younger than 7 years; therefore, it is not a good screening test for diagnosis. RF positivity is more common in children who have disease onset at a later age, polyarticular disease, or subcutaneous rheumatoid nodules. Seropositivity is associated with erosive synovitis and poorer prognosis. HLA-B27 is not diagnostic but is associated with reactive arthritis, IBD, and ERA.4,6
Analysis of synovial fluid
Synovial fluid analysis and culture should be performed in children with acute joint swelling and pain accompanied by fever or for whom the diagnosis is uncertain or an infectious cause is suspected. Gout and pseudogout are extremely rare in children and only under the most unusual circumstances does synovial fluid need to be sent for crystal analysis. Synovial fluid should be analyzed at the time of an intra-articular corticosteroid injection, if possible. In JIA, the synovial fluid usually is yellow and cloudy, with decreased viscosity.
Leukocyte counts are elevated (typically between 15,000 and 20,000/µL, but may be higher), with neutrophil predominance. Synovial fluid leukocyte counts of 2000 to 50,000/µL are defined as inflammatory; with higher counts, infection needs to be considered.
Imaging
Radiography typically is best for the initial evaluation of arthritis to look for juxta-articular osteopenia, erosions, growth-arrest lines, fractures, and other bony abnormalities, but these abnormalities may not be apparent for several months. Among children with JIA, radiographic features include, in order of progression, soft tissue swelling, widening or narrowing of the joint space, osteoporosis, erosions, subluxation, and ankylosis. Erosive changes are uncommon before 2 years of active disease.
Other useful imaging modalities include ultrasonography, bone scanning, and MRI. Ultrasonography is a noninvasive and inexpensive method to confirm a joint effusion. Bone scans help detect osteomyelitis, malignancy, and joints that have subclinical inflammation. MRI is the most sensitive technique for identifying soft tissue abnormalities and early erosions. It is especially useful in looking at joints otherwise difficult to evaluate, such as the temporomandibular (TMJ), hip, and sacroiliac joints, as well as at TMJ arthritis.4 For children with JIA, early disease identification and treatment leads to improved quality of life.
References:
References1. Hayward K, Wallace CA. Recent developments in anti-rheumatic drugs in pediatrics: treatment of juvenile idiopathic arthritis. Arthritis Res Ther. 2009;11:216.
2. Wallace CA. Current management of juvenile idiopathic arthritis. Best Pract Res Clin Rheumatol. 2006;20:279-300.
3. Sullivan KE. Inflammation in juvenile idiopathic arthritis. Rheum Dis Clin North Am. 2007;33:365-388.
4. Cassidy JT, Petty RE. Chronic arthritis in childhood. In: Cassidy JT, Petty RE, Laxer RM, Lindsley CB, eds. Textbook of Pediatric Rheumatology. 5th ed. Philadelphia: Elsevier Saunders. 2005:206-260.
5. Weiss JE, Ilowite NT. Juvenile idiopathic arthritis. Rheum Dis Clin North Am. 2007;33:441-470, vi.
6. Woo P, Laxer RM, Sherry DD. Juvenile idiopathic arthritis. Pediatric Rheumatology in Clinical Practice. London: Springer-Verlag; 2007:23-46.
7. Schneider R, Passo MH. Juvenile rheumatoid arthritis. Rheum Dis Clin North Am. 2002;28:503-530.
8. Petty RE, Southwood TR, Manners P, et al; International League of Associations for Rheumatology. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton 2001. J Rheumatol. 2004;31:390-392.














