Managing Neuropsychiatric Lupus: Top 10 Clinical Pearls
Once a diagnosis of systemic lupus erythematosus (SLE) has been made, numerous neurological and psychiatric manifestations may be found to accompany the disease.
Once a diagnosis of systemic lupus erythematosus (SLE) has been made, numerous neurological and psychiatric manifestations may be found to accompany the disease. CNS involvement in patients with SLE has been documented for more than 100 years,1 but the many signs and symptoms of neuropsychiatric lupus (NPSLE) continue to present primary care physicians and specialists with a significant diagnostic and therapeutic challenge. Heterogeneous brain symptoms (eg, cognitive impairment, psychosis, mood or anxiety disorder, and intractable headache) are difficult to attribute to NPSLE without validated biomarkers. NPSLE has well-documented morbidity and mortality resulting from SLE-related CNS involvement, including suicide, but they may be prevented if physicians take a more proactive approach to patient care.
In this article, we attempt to simplify the difficult and controversial tasks of diagnosis and management of NPSLE by describing a clinical approach to patients where evidence-based medicine is lacking. We discuss the pathogenesis, diagnosis, and various levels of treatment.
We offer 2 cases to demonstrate the success of appropriate evaluation with long-term treatment and our “top 10 clinical pearls,” which have helped us approach NPSLE with a proactive strategy for 3 decades. Our goals are to demystify NPSLE, encourage more physicians to take a proactive approach to this complex problem, and prevent the morbidity and mortality that may result from underdiagnosis and undertreatment.



Clinical presentations
Although diagnosis of NPSLE is one of the most difficult areas of medicine, progress is being made. Determination of the neuropsychiatric symptoms in SLE by American College of Rheumatology (ACR) nomenclature has helped standardize clinical presentations in research and clinical practice (Table 1).2 This nomenclature also has been incorporated into the British Isles Lupus Assessment Group (BILAG) 2004 index to standardize clinical trials with SLE and help therapeutic decision making.3
Pathogenesis



What makes NPSLE so complex and heterogeneous-with 19 case definitions-is the multiplicity of proposed pathogenic mechanisms. Three mechanisms are currently described: autoantibodies, vascular abnormalities, and inflammatory mediators (Table 2).4
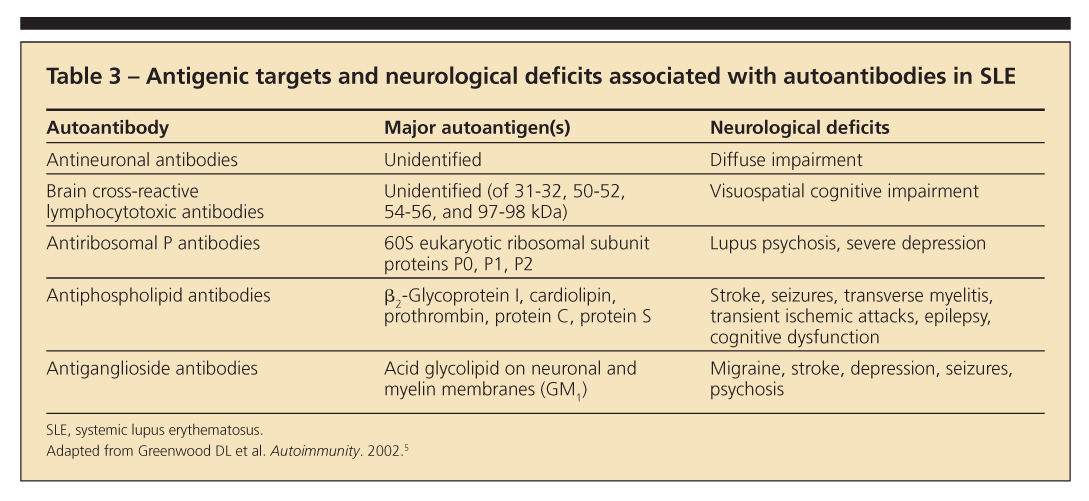
All of these autoantibodies are helpful when used with other clinical data (Table 3).5 However, studies of associations looked at individually have been inconclusive.
The role of antineuronal anti–N-methyl D-aspartate (NMDA) recepto


r antibodies is the most recent example. In 2008, a published paper suggested that NMDA antibodies in cerebrospinal fluid (CSF) are correlated with diffuse NPSLE but not those in serum.6 However, an abstract published later in the year did not confirm these data.7
Antibodies to microtubule-associated protein were found in patients with NPSLE more than in patients who had SLE with no NPSLE, patients with multiple sclerosis (MS), and healthy controls.8 Although these microtubular antibodies are important for further research, they are not currently available for clinical practice.
B-cell–depleting therapies currently are being evaluated in SLE. An important study looked at B-lymphocyte stimulator (BLyS), an inflammatory cytokine that modulates B-cell activity.9 Increased levels of this cytokine were found that correlated with clinical activity of SLE; on serial visits, the BLyS level increased when there was increased lupus activity.
The vascular abnormalities of NPSLE involve an activated vascular endothelium, leading to a noninflammatory vasculopathy in the presence of antiphospholipid (aPL) antibodies, and leukothrombosis in the presence of complement or endothelial activation.5 On a clinical level, these intravascular processes usually are not correlated with the classic signs or laboratory tests of SLE systemic activity (eg, erythrocyte sedimentation rate and C-reactive protein, serum complement, and double-stranded DNA levels). In a speculative model for NPSLE, antibodies can cross the blood-brain barrier and cause neuronal damage because the ongoing vasculopathy leads to disruption of this barrier.5
A true inflammatory vasculitis is very rare in NPSLE, and the presence of vasculitis should prompt the clinician to consider another diagnosis, such as primary CNS vasculitis. Although rare, reversible cerebral vasoconstriction syndromes mimic CNS vasculitis and should be considered to avoid unnecessary treatment with corticosteroids, cyclophosphamide (CYC), or biologic agents.10 Other inflammatory cytokines (eg, interleukin-6) are present in the CSF with active NPSLE when correlating serum levels are normal.11
Genetic markers for disease activity in NPSLE currently are being evaluated in the interferon-α response pathway as measured by gene expression. This expression may serve as a molecular diagnostic tool to determine active inflammatory changes causing NPSLE that require more intense therapy with drugs discussed in this article.
Diagnosis
The diagnosis of NPSLE can be made only on a case-by-case basis. ACR case definitions, select autoantibodies, CSF analysis, neuroimaging, and neuropsychological testing may be used.12 Infection always is the major differential concern; evaluation should include blood cultures, CSF culture, or brain or spine neuroimaging techniques.
Corroboration of concomitant use of other medications and any substance abuse should be sought from a third party who knows the patient well. If clinically indicated, echocardiograms and carotid Doppler ultrasonograms should be obtained to rule out atheroembolic disease to the brain, which is accelerated in active SLE.
The most difficult part is differentiating organic disease from functional disease. A battery of neuropsychological tests validated by the ACR are considered reliable. More comprehensive testing may be useful for identifying specific deficits in NPSLE.13
Although multiple types of sophisticated neuroimaging have been proposed for NPSLE, standard brain or spine MRI is still the modality of choice for detecting and monitoring vascular ischemic lesions and demyelinating lesions. In a recent study of patients who had a new diagnosis of SLE, 25% had brain abnormalities on MRI scans.14 Although these patients will be monitored for reversibility and progression, clinical correlation must be made with MRI and clinical changes in individual patients. Brain MRI changes can be evanescent regardless of treatment. Clinical correlation prevents treatment of insignificant MRI changes.
Progressive focal strokes (cerebrovascular disease by ACR case definitions) may correlate with progressive focal lesions in subcortical white matter on brain MRI scanning. With this presentation, a patient who has known aPL antibodies correlates pathogenic mechanism, clinical progression, and MRI changes. In this clinical setting, anticoagulation would be the mainstay of initial treatment. Encephalopathic neuropsychiatric manifestations of SLE often have no associated visible lesions on MRI.
CSF analysis rules out infection, but the results may be normal in NPSLE. Elevated protein levels are found in only up to half of patients, and pleocytosis (white blood cell count higher than 5/µL) is seen in up to one-third of patients.15 The CSF IgG synthesis rate may be elevated and oligoclonal banding may be present in up to 80% of patients, especially those who have associated CNS demyelination and clinical MS or lupoid sclerosis.
Treatment (conventional approaches)
Because NPSLE has so many case definitions that occur in various combinations and, possibly, different pathogenic mech



anisms, treatment should be tailored for each patient. Once infection and metabolic and hypertension syndromes are ruled out, symptomatic therapies are started (Table 4).4 They include anticonvulsants, axiolytics, antidepressants, psychotropics, antimigraine medicines, and treatments for secondary fibromyalgia syndrome (FMS).
In progressive cases, therapy should not be delayed pending test results. Most clinicians recommend 1 mg/kg/d of corticosteroids in divided doses or, in more severe cases, pulse IV methylprednisolone, 0.5 to 1 g/d for 3 days. At this point, with continued clinical progression, early, aggressive management and corticosteroid tapering have led to a better prognosis in patients with SLE.16
Our group and others have shown IV CYC in doses of 0.5 to 1 g/m2 to be beneficial (see “Recommended readings.”). In the most severe cases, with the patient in a coma or with acute transverse myelitis, many centers suggest plasmapheresis.17 At all levels of progression, anticoagulation is paramount if thrombosis is one of the pathogenic mechanisms.


Figure 1 –A white woman with neuropsychiatric systemic lupus erythematosus (NPSLE), currently age 50 years, first presented in 2002. She was switched from intravenous cyclophosphamide (IV CYC) to rituximab in 2003. In April 2005, she had a severe flare of NPSLE with tremor, a 4-day amnesic event, and significant memory decline. The accompanying brain MRI scan (A) showed progression of her disease with 13 lesions (arrows). IV CYC was added to rituximab in combination, and by July 2005, a second MRI scan (B) showed a reduction to 6 lesions with dramatic clinical improvement. The results of yearly MRI scans obtained through 2009 have remained stable, and the patient’s clinical status has been unremarkable with the combination treatment of IV CYC and rituximab every 6 months.


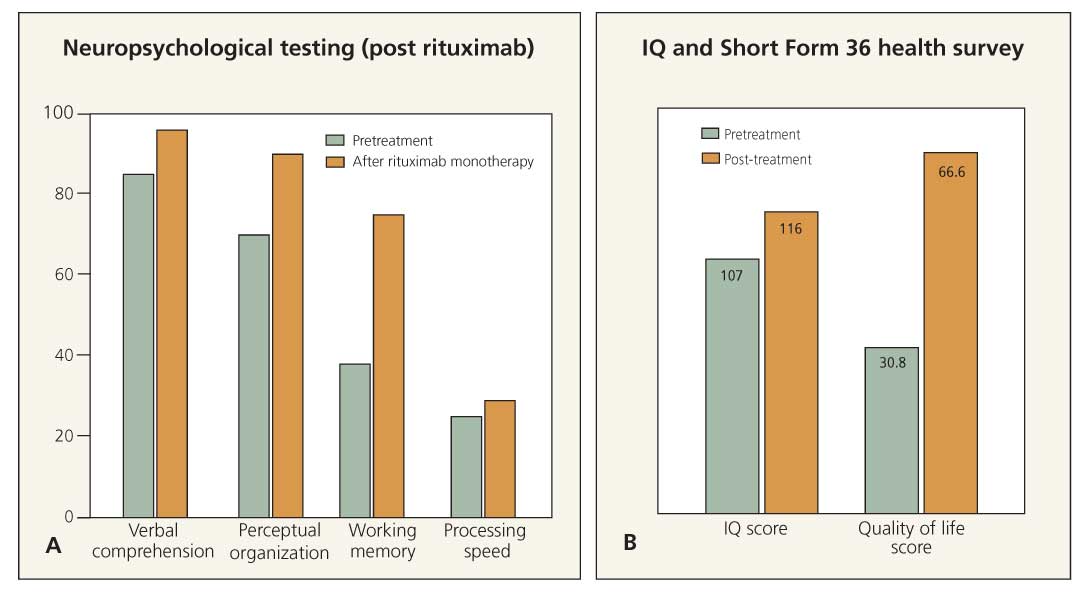
Figure 2 –A white woman who received a diagnosis of neuropsychiatric systemic lupus erythematosus in 2000 is currently age 46 years. In 2001, she had continuing auditory and visual hallucinations with acute confusional episodes. In 2003, her first neuropsychological testing showed significant defects in verbal comprehension, perceptual organization, working memory, and processing speed. Intravenous cyclophosphamide (IV CYC) was no longer effective, and she was started on a regimen of rituximab therapy in November 2003 at 6-month intervals. In June 2005, repeated neuropsychological testing showed improvement in all of the above parameters (A). Her IQ improved from 107 to 116 on the Wechsler Adult Intelligence Scale, and the Short Form 36 health survey showed that her quality of life improved from 30.8 to 66.6 (B). The patient subsequently has required IV CYC/rituximab combination therapy for ongoing CNS events. In 2009, she has maintained her quality of life.
In cases of progressive disease in which more aggressive treatment with corticosteroids or immunosuppressive agents should be initiated, consultation with a physician experienced in managing progressive NPSLE may be helpful. Determining whether aggressive treatment is working can be answered by following objective parameters, such as CSF changes, brain MRI changes (Figure 1), and serial neuropsychological testing (Figure 2).
Treatment (novel approaches)
In select refractory cases, the use of intrathecal methotrexate with dexamethasone has shown a 90% response rate.18 Because current therapies are associated with treatment-limiting toxicities-including cytopenia, infections, ovarian failure, secondary malignancy, and osteoporosis with continued corticosteroid use-new options are needed.
Rituximab, a targeted monoclonal antibody against CD20+ B cells, was studied in SLE and did not meet its end points.19 However, the study excluded patients with severe NPSLE and included therapy with other immunosuppressive drugs. Open-label efficacy and safety has been shown with rituximab in refractory NPSLE and continues to hold promise. Placebo-controlled, double-blinded trials of rituximab in combination with IV CYC currently are being proposed. Validation for these studies is supported by a 72% response rate in open-label selective data when conventional corticosteroids and IV CYC were not successful.20
In the future, treatment end points will be based on the BILAG 2004 index, the Systemic Lupus Erythematosus Disease Activity Index 2000, and the Physician’s Global Assessment system. We currently rely on select open-label data on the use of rituximab in severe, progressive NPSLE when conventional therapy has not been successful. Two isolated cases show how current tools may be used to monitor treatment response to novel treatment protocols (see Figures 1 and 2).
Top 10 clinical pearls
Good prospective data will emerge eventually. For now, we offer our top 10 pearls to provide practical recommendations for treating patients in office practice.
1. The answers to key questions asked of patients may suggest NPSLE rather than depression, FMS, or somatoform pain disorder. Patients will not answer voluntarily unless the questions are gentle but probing.
• “Do you have difficulty in reading and retaining what you read?” Professionals (eg, doctors, nurses, lawyers, business executives) often break down when asked and state, “No one has ever asked me that before.” They are concerned that their careers could be in jeopardy.
• “Do you ever see people or animals that are not there or hear voices?” Visual hallucinations, particularly those associated with some insight into not being able to think correctly, are characteristic of the organic psychosis we have observed in NPSLE. Hallucinations related to drug or substance abuse should be excluded. Most patients are reluctant to reveal this information, fearing that they will be labeled “crazy.” When asked gently, patients are relieved to reveal their secret and hopeful when told that there may be specific treatment. Patients feel validated when told that these psychotic episodes may be part of the NPSLE spectrum.
2. Cognitive impairment is common in patients with SLE and may reach the severity range of dementia. The pattern is different from that of other dementias, such as Alzheimer disease. Characteristically affected are executive function, concentration, thought-processing speed, and word finding; however, vocabulary is preserved so that in normal conversation, the deficit may not be apparent. Patients should be identified with serial neuropsychological testing. If significant worsening is found, cognitive or medicinal treatment should be instituted. The exact therapeutic regimen to be used is unclear; treatment is based on an identifiable pathogenic mechanism (eg, aPL) or other presenting signs (eg, psychosis, seizures).
3. When serial neurocognitive testing is not possible, the cognitive dysfunction should be sufficiently severe to interfere with daily activities and, if possible, a corroborating history from a third party should be obtained, as stated in BILAG 2004, to establish the diagnosis of NPSLE. Input from spouses, family members, or friends is almost imperative in establishing the diagnosis and measuring response to treatment. Some patients with significant NPSLE have difficulty in remembering or quantifying their own improvement.
4. All patients with NPSLE should undergo screening tests for aPL. If focal neurological deficits correlate with positive antibodies, anticoagulation may be the mainstay of treatment and prevent overuse of corticosteroids, IV CYC, or B-cell–depleting agents. NPSLE also may present as MS or with apparent demyelinating lesions on MRI in the absence of clinical MS. In the presence of aPL, these presentations may instead represent ischemic demyelination.
5. Movement disorders that have been noted in CNS lupus (eg, atypical tremors, chorea, tics, and extrapyramidal syndromes) may improve with management of the underlying disease. Treatment is symptomatic in the same way as in other, similar movement disorders, but the response may not be as good in the absence of managing the underlying lupus.
6. Rheumatologists have adopted an early and aggressive management approach to rheumatoid arthritis, and this approach may be a contributor to increasing the survival of patients with lupus.16 A similar approach should be adopted in NPSLE, because when brain function is reduced, a patient may not be able to work or read, and this may result in a major decline in quality of life. Research with biologic drug trials has led to new treatment paradigms as in MS, which can manifest in CNS lupus. Early and aggressive management of MS, while the disease is still in the inflammatory phase, was suggested in a recent editorial.21
7. All patients with progressive neurocognitive testing or progressive brain MRI scanning results correlating with clinical signs and symptoms of NPSLE should undergo CSF analysis. The results may be abnormal and help substantiate a clinician’s clinical diagnosis with another objective parameter. The more objective parameters found in individual patients, the greater the confidence physicians may have in the diagnosis and treatment plan. CSF studies should involve cells, protein, IgG synthesis rate, oligoclonal banding, antineuronal antibodies, antiribosomal P antibodies, and culture. The JC virus detected by DNA polymerase also should be included to rule out progressive multifocal leukoencephalopathy, usually in patients being treated with immunosuppressive or immunomodulatory drugs.
8. In formulating treatment plans for the most severe cases, make sure that the patient and family and friends understand the risk/benefit ratio and agree with the plan. Shared decision making can allay concerns about drug toxicity and adverse effects and encourage compliance with treatment.
9. Management of NPSLE requires a team approach, including the rheumatologist, neurologist, psychiatrist, neuropsychologist, oncologist, and primary care physician, to formulate a proper diagnosis and treatment plan. Because there is no “gold standard” diagnostic test or biopsy, both overdiagnosis and underdiagnosis are inevitable; this varies with the confidence and experience physicians have in dealing with NPSLE.
10. Even if a clinician has vast experience, there will be some degree of uncertainty. If a proactive approach is taken, clinical judgment usually leads to appropriate diagnosis and treatment in most patients. The need for appropriate treatment is substantiated by prospective data that show that NPSLE is associated with a poor clinical outcome, recurrence, and a 10% chance of death resulting from the NPSLE involvement. Despite advances in treatment since 1950, organ damage in NPSLE has not been reduced, and concerns have been raised that total damage in NPSLE will continue.22
References:
References1. Osler W. On the visceral complications of erythema exudativum multiforme. Am J Med Sci.1895;110:629-646.
2. American College of Rheumatology Ad Hoc Committee on Neuropsychiatric Lupus Nomenclature. The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheum. 1999;42:599-608.
3. Yee CS, Farewell V, Isenberg DA, et al. British Isles Lupus Assessment Group 2004 index is valid for assessment of disease activity in systemic lupus erythematosus. Arthritis Rheum. 2007;56:4113-4119.
4. Hanly JG. Neuropsychiatric lupus. Rheum Dis Clin North Am. 2005;31:273-298, vi.
5. Greenwood DL, Gitlits VM, Alderuccio F, et al. Autoantibodies in neuropsychiatric lupus. Autoimmunity. 2002;35:79-86.
6. Arinuma Y, Yanagida T, Hirohata S. Association of cerebrospinal fluid anti-NR2 glutamate receptor antibodies with diffuse neuropsychiatric systemic lupus erythematosus. Arthritis Rheum. 2008;58:1130-1135.
7. Fragoso-Loyo HE, Cabiedes J, Orozco-Narvaez A, et al. Paired analysis of autoantibodies, cytokines and chemokines in cerebrospinal fluid (CSF) of SLE patients with central neuropsychiatric manifestations (cNPSLE) with and without associated factors. Arthritis Rheum. 2008;58(suppl):S636.
8. Lefranc D, Launay D, Dubucquoi S, et al. Characterization of discriminant human brain antigenic targets in neuropsychiatric systemic lupus erythematosus using an immunoproteomic approach. Arthritis Rheum. 2007;56:3420-3432.
9. Petri M, Stohl W, Chatham W, et al. Association of plasma B lymphocyte stimulator levels and disease activity in systemic lupus erythematosus. Arthritis Rheum. 2008;58:2453-2459.
10. Singhal AB, Kimberly WT, Schaefer PW, Hedley-Whyte ET. Case records of the Massachusetts General Hospital. Case 8-2009: a 36-year-old woman with headache, hypertension, and seizure 2 weeks post partum. N Engl J Med. 2009;360:1126-1137.
11. Hirohata S, Miyamoto T. Elevated levels of interleukin-6 in cerebrospinal fluid from patients with systemic lupus erythematosus and central nervous system involvement. Arthritis Rheum.1990;33:644-649.
12. Hanly JG. New insights into central nervous system lupus: a clinical perspective. Curr Rheumatol Rep. 2007;9:116-124.
13. Kozora E, Ellison MC, West S. Reliability and validity of the proposed American College of Rheumatology neuropsychological battery for systemic lupus erythematosus. Arthritis Rheum. 2004;51:810-818.
14. Petri M, Naqibuddin M, Carson KA, et al. Brain magnetic resonance imaging in newly diagnosed systemic lupus erythematosus. J Rheumatol. 2008;35:2348-2354.
15. West SG. The nervous system. In: Wallace DJ, Hahn BH, eds. Dubois’ Lupus Erythematosus. 7th ed. Baltimore: Lippincott Williams & Williams; 2007:731.
16. Nived O, Sturfelt G, Bengtsson AA. Improved lupus outcome: we are doing a good job, but could we do better? J Rheumatol. 2008;35:2088-2089.
17. Bartolucci P, Bréchignac S, Cohen P, et al. Adjunctive plasma exchanges to treat neuropsychiatric lupus: a retrospective study on 10 patients. Lupus. 2007;16:817-822.
18. Valesini G, Priori R, Francia A, et al. Central nervous system involvement in systemic lupus erythematosus: a new therapeutic approach with intrathecal dexamethasone and methotrexate. Springer Semin Immunopathol. 1994;16:313-321.
19. Merrill JT, Neuwelt CM, Wallace DJ, et al. Efficacy and safety of rituximab in patients with moderately to severely active systemic lupus erythematosus (SLE): results from the randomized, double-blind phase II/III study EXPLORER. Arthritis Rheum. 2008;58(suppl). Late-breaking abstract.
20. Neuwelt CM, Young RG, Daikh DI. Comparison of rituximab or cyclophosphamide monotherapy with combined rituximab/cyclophosphamide for severe central nervous system lupus erythematosus. The 8th International Congress on SLE; 2007; Shanghai.
21. Hauser SL. Multiple lessons for multiple sclerosis. N Engl J Med. 2008;359:1838-1841.
22. Kantharajpur S, Petri M. Increase in neuropsychiatric and total organ damage in SLE since 1950 1979. Arthritis Rheum. 2008;58(suppl):S330-S331.
Recommended readings
The authors suggest the following sources for readers seeking more information:
• Barile-Fabris L, Ariza-Andraca R, OlguÃn-Ortega L, et al. Controlled clinical trial of IV cyclophosphamide versus IV methylprednisolone in severe neurological manifestations in systemic lupus erythematosus. Ann Rheum Dis. 2005;64:620-625. This paper presents the only controlled trial that compares cyclophosphamide and corticosteroids taken intravenously.
• Calabrese LH, Molloy ES, Huang D, Ransohoff RM. Progressive multifocal leukoencephalopathy in rheumatic diseases: evolving clinical and pathological patterns of disease. Arthritis Rheum. 2007;56:2116-2128. Although many of the protean manifestations of progressive multifocal leukoencephalopathy (PML) may be difficult to distinguish from those of neuropsychiatric systemic lupus erythematosus (NPSLE), many of the cases of PML in SLE occur with minimal immunosuppression.
• Denburg SD, Carbotte RM, Denburg JA. Corticosteroids and neuropsychological functioning in patients with systemic lupus erythematosus. Arthritis Rheum. 1994;37:1311-1320. This paper presents the only controlled trial of corticosteroids in managing NPSLE.
• Neuwelt CM, Lacks S, Kaye BR, et al. Role of intravenous cyclophosphamide in the treatment of severe neuropsychiatric systemic lupus erythematosus. Am J Med. 1995;98:32-41. Although a retrospective review of 31 patients with neuropsychiatric lupus who were treated with intravenous cyclophosphamides and for whom previous therapies had not succeeded, including corticosteroids and other immunosuppressives, this paper emphasized clinical presentation and response to treatment.
• Tokunaga M, Saito K, Kawabata D, et al. Efficacy of rituximab (anti-CD20) for refractory systemic lupus erythematosus involving the central nervous system. Ann Rheum Dis. 2007;66:470-475. Rituximab rapidly improved refractory CNS lupus, as evidenced by resolution of various clinical signs and symptoms and improvement of radiographic signs.















