Granulomatosis With Polyangiitis, With Atypical Features
The manifestations of granulomatosis with polyangiitis varies widely. This case study describes a 49-year-old man who presented with cough, chills, and chest pain who had a mediastinal mass and ocular involvement.
A 49-year-old uninsured man from Mexico who had hypertension, diabetes and chronic rhinitis and sinusitis presented for hospitalization with two weeks of cough and hemoptysis, culminating in two days of chills, night sweats, left-sided chest pain that radiated to the shoulders, as well as dyspnea on exertion and orthopnea.


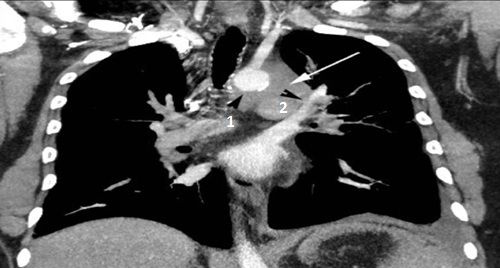
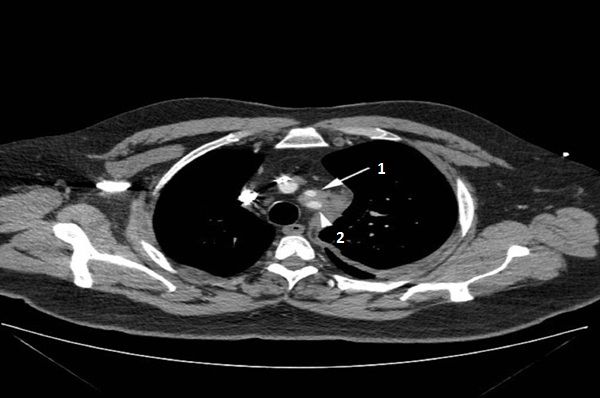
A chest CT with contrast showed an 8.5 cm left mediastinal mass extending to the left hilum (above, white arrow), abutting the aorta (above, black arrow head 1), main pulmonary artery, and left pulmonary artery (above, black arrow head 2), encasing the left subclavian artery (below, arrow head 2) and origin of the left common carotid artery (below, white arrow 1).


Two years ago, polyarthritis developed in his shoulders, wrists, hands, knees, and ankles. A year later, microscopic hematuria and proteinuria were noted, with normal creatinine clearance.
Six months previously, a pacemaker had been implanted for complete heart block, after he presented with erythema of the left eye, pain, photophobia, headaches, and the sensation that his eyes were “popping out.” Our review of a chest CT taken at that time showed a large pericardial effusion, pulmonary vascular congestion with patchy ground-glass opacities bilaterally, interstitial infiltrates, pneumonitis in the right lower lobe, and a small left mediastinal mass.
Highest on the list for differential diagnosis were neoplastic, infectious, and inflammatory causes. We performed an infectious workup, a lung biopsy, and autoimmune serologies.
Click the link at right to read the clinical findings.


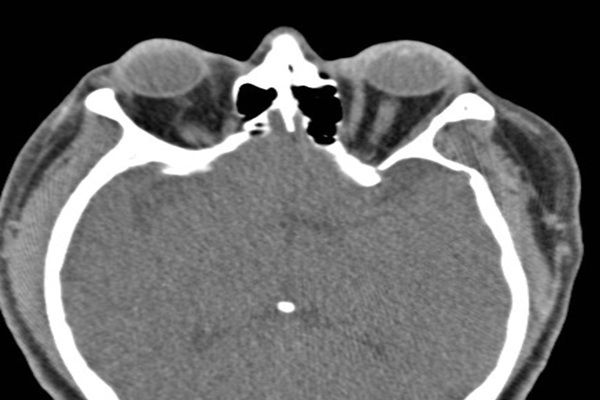
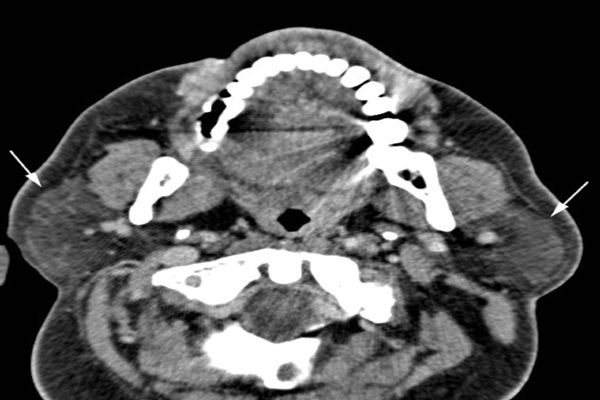
CT of the head and orbits showed normal sinuses, bilateral proptosis with increased caliber of the inferior and medial rectus muscles bilaterally (left), and bilateral parotid gland enlargement (below, white arrows). Ophthalmology diagnosed left pan-uveitis and neuroretinitis, which after a negative infectious work-up was thought to be possible vasculitis.
Serologies revealed a C-ANCA of 1:320 and PR3 antibody of 28.5.
The overall clinical picture was consistent with an ANCA-associated systemic vasculitis with both typical (sinus, lung, articular, and renal) and atypical (orbital, parotid, pericardial, conduction system, and mediastinal) features. We diagnosed granulomatosis with polyangiitis (GPA).

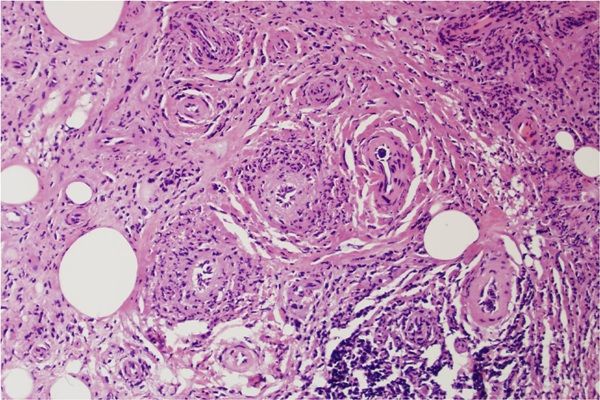
VATS biopsy of the left lung mass revealed vasculitis involving the arterioles with resultant angiodestruction, relative sparing of the small arteries, and numerous necrobiotic and angiocentric granulomas (below). Marked fibrosis was present, with positive immunofluorescence for IgG and fibrin within the arterioles, consistent with GPA.

Once we had obtained biopsy confirmation, the patient began treatment with the RITUXIVAS protocol,4 followed by a prednisone taper.
Continue to theDiscussion.
Discussion: ANCA-associated vaculitides, including GPA (formerly called Wegener’s granulomatosis), microscopic polyangiitis, and the Churg-Strauss syndrome, are identified by the presence of antineutrophil cytoplasmic antibodies that can be specific for the myeloperoxidase or proteinase 3 cytoplasmic antigens. These activate neutrophils and monocytes, leading to damage to the vascular endothelium of small to medium-sized vessels.
The presentation of these diseases varies so greatly between individuals that genetic, epigenetic, and environmental factors are suspected to play an important role in determining the phenotype. The lungs are involved in up to 90% of cases, and there is renal involvement in about 70-80%.1,4
Pulmonary manifestations in GPA are varied and can mimic pneumonia, neoplasm, and other noninfectious inflammatory diseases. Most commonly seen in GPA are multiple lung nodules, in about 40-70%, and infiltrates in up to 50%. About 25-50% of these progress to cavitary lesions, some of which can become secondarily infected. Sometimes hemorrhage can occur around these lesions; in about 30% of cases, they lead to lung consolidation and ground-glass opacities, which can arise as a result of alveolar hemorrhage, cellular infiltrates, or small-vessel vaculitis. Diffuse alveolar hemorrhage occurs in about 10%.
Other manifestations include pleural disease and occasionally mediastinal masses, as in our patient.1-3
For nearly the last four decades, the standard remission induction therapy has involved treatment with cyclophosphamide and high-dose glucocorticoids. This regimen is effective in 70-90% of cases, but can be associated with severe toxicity, such as cytopenias, infertility, infections, bladder injury, and cancer, along with adverse effects from chronic glucocorticoid use.
Disease activity in ANCA-associated vasculitis correlates with the percentage of activated peripheral B lymphocytes , and cyclophosphamide suppresses the activation, proliferation, and differentiation of autoreactive B cells. More recently, sustained remissions in refractory ANCA-associated vasculitis have been reported in 80-90% of patients treated with rituximab.4 An anti-CD20 monoclonal antibody, rituximab depletes B cells that produce antibodies, such as the ANCAs that have an affinity for the proteinase-3 antigen.
Two landmark randomized trials, RITUXIVAS4 and RAVE,5 specifically investigated the effects of anti-B-cell induction therapy with rituximab head-to-head against cyclophosphamide in patients with severe ANCA-associated vasculitis. The findings were similar: Essentially, rituximab was as effective in inducing remission and was not inferior to cyclophosphamide, delivered in either intravenous (RITUXIVAS) or oral forms (RAVE).
However, the patients receiving rituximab in the RITUXIVAS trial also received at least two doses of intravenous cyclophosphamide, whereas in the RAVE trail they did not receive any cyclophosphamide. In both trials, the cyclophosphamide groups received maintenance therapy with azathioprine, whereas the rituximab treatment groups did not receive any specific maintenance therapy. It is still unknown whether maintenance therapy should play a role in the rituximab regimen after peripheral B cells return, as expected, about 9 to 12 months following the end of treatment.
Those receiving rituximab did have a significant decrease in ANCAs in both trials, but more so in the RITUXIVAS trial, where no patients had a positive ANCA 6 months after induction. ANCA negativity after induction has been associated with a reduced risk of relapse.6 In addition, the RAVE trial’s subgroup analysis showed that rituximab was superior to oral cyclophosphamide for the induction of remission in relapsing disease.
Long-term safety differences between the two regimens for ANCA-associated vasculitis have yet to be established. In rheumatoid arthritis and non-Hodgkin’s lymphoma, rituximab has not been associated with significant increases in infectious complications even when used with other DMARDs or with chemotherapy, and unlike cyclophosphamide it rarely results in leukopenia. It had been hoped that the rituximab groups in RITUXIVAS and RAVE would show significant reductions in adverse events. Some adverse events were disease- or steroid- related, so it remains unclear whether and to what degree either cyclophosphamide or rituximab contribute to the burden of adverse effects.
Long-term data regarding relapses beyond 6 to 12 months for these regimens may reveal whether cyclophosphamide should be used in conjunction with rituximab for a more relapse-free, sustained remission with fewer overall adverse events. We may also learn more about the role of rituximab as maintenance therapy in ANCA-associated vasculitis.
Outcome: With our uninsured patient about to receive his first induction therapy for severe GPA, with likely renal involvement, we made the collective decision to proceed with the shortest and most relevant induction regimen from the RITUXIVAS trial.4 This allowed him to have reduced exposure to cyclophosphamide and to avoid maintenance immunosuppression other than glucocorticoids.
He showed significant initial improvement in his arthritis, eye pain, headaches, and respiratory symptoms. Unfortunately, we lost him to follow-up after two months, because he decided to return to Mexico.
(Scroll below the Comments box for the References.)
References:
REFERENCES:
1. Ananthakrishnan L, Sharma N, Kanne JP. Wegener’s Granulomatosis in the Chest: High-Resolution CT findings. AJR (2009) 192:676–682.
2. Cordier JF, Valeyre D, Guillevin L et al. Pulmonary Wegener’s Granulomatosis. A Clinical and Imaging Study of 77 Cases. Chest (1990) 97:906-912.
3. Maguire R, Fauci AS, Doppman JL, and Wolff SM. Unusual Radiographic Features of Wegener’s Granulomatosis. Am J Roentgenol (1978) February:130233-130238.
4. Jones RB, Tervaert JWC, Hauser T, et al for the European Vasculitis Study Group. Rituximab versus Cyclophosphamide in ANCA-Associated Renal Vasculitis. N Engl J Med (2010) 363:211-220.
5. Stone JH, Merkel PA, Spiera R, et al for the RAVE-ITN Research Group. Rituximab versus Cyclophosphamide for ANCA-Associated Vasculitis.N Engl J Med (2010)363:221-32.
6. Sanders JS, Huitma MG, Kallenberg CG, and Stegeman CA. Prediction of relapses in PR3-ANCA-associated vasculitis by assessing responses of ANCA titres to treatment.Rheumatology (Oxford) (2006)45:724-9.














