Identifying and Managing Dermatomyositis: A Case Report and Review
This detailed review desccribes the heterogeneous idiopathic inflammatory myopathies and their mimics, with criteria for classification, an overview of cardiac, pulmonary and malignant comorbidieis, and guidance for treatment.
ABSTRACT: The idiopathic inflammatory myopathies are a heterogeneous group of disorders; excluding conditions that mimic them during the initial patient evaluation is essential. Classification of dermatomyositis (DM) for a definitive diagnosis requires a characteristic rash and other criteria, such as proximal muscle weakness and muscle enzyme level elevation. DM often overlaps with other connective tissue diseases. A photosensitive rash often is the initial manifestation. Cardiac and pulmonary involvement may be life-threatening. Malignancies occur in up to 25% of cases. A detailed strength examination at every visit is important for assessing treatment response. Muscle biopsy is central in establishing the diagnosis. Physical therapy and occupational therapy should be started at diagnosis. Corticosteroids are the foundation of treatment. Most patients require corticosteroid-sparing medication.
(J Musculoskel Med. 2008;25:415-420, 444)
Dermatomyositis (DM) is one of the idiopathic inflammatory myopathies (IIMs), a heterogeneous group of disorders that involve proximal muscle weakness and nonsuppurative skeletal muscle inflammation. Others are polymyositis (PM) and inclusion body myositis (IBM). Excluding conditions that mimic the IIMs during the initial patient evaluation is essential.1 Not every patient who has myopathic weakness, elevated creatine kinase (CK) levels, and inflammatory muscle histology has DM or PM, and there is clinical and pathological overlap with myositis associated with collagen vascular diseases. In addition, other conditions that do not respond to immunosuppression (eg, muscular dystrophy, IBM, metabolic myopathies, and neuromuscular disorders) also may have these features.
If treatment of a patient with DM or PM is unsuccessful, the muscle biopsy should be reexamined or a second biopsy ordered to reassess the diagnosis. Elevated levels of aspartate aminotransferase (AST), alanine aminotransferase (ALT), and lactate dehydrogenase (LDH) in patients with nonspecific symptoms often direct attention to liver disease. These enzymes also are found in muscle; if levels are elevated, the CK level should be checked to evaluate for myositis. Although the CK level is helpful, the response to therapy is best appraised by monitoring muscle strength and function. "Treating" the CK level instead of the muscle weakness may lead to unnecessarily prolonged use of immunosuppressive medications and incorrect judgment about their efficacy.2
In this article, we offer a case report of a patient with DM. Then we review the key points of diagnosis and treatment.
CASE
A 39-year-old woman, in her eighth week of pregnancy, was admitted with myalgia, proximal muscle weakness, and a rash that had developed over the course of 1 week. She experienced aching pain that worsened with activity; thigh and proximal arm muscle fatigue and weakness began 3 days later. Then a mildly pruritic rash across the forehead, nose, and forearms developed. Her symptoms progressed rapidly, and within days she required assistance from 2 persons to rise from a chair; she also had difficulty walking, brushing her teeth, bringing objects to her mouth, and washing her hair. She felt feverish (temperature, 37.2°C [99°F]).
One year earlier, about 3 to 4 weeks into a pregnancy, the patient had experienced similar proximal myalgia and very mild weakness without rash. The symptoms had resolved in 2 weeks without medical attention. This pregnancy ended in miscarriage at 10 weeks.
The patient smoked cigarettes, 1 pack a day for 15 years, but she denied alcohol or illicit drug use. The only medication she took was a prenatal vitamin.
Physical examination revealed a slight periorbital heliotrope rash around the eyelid margin and erythematous plaques with a thin scale overlying the forehead, scalp line, nose, cheeks, neck, and anterior chest (Figure 1). Similar plaques, in a linear pattern, were observed on the forearms (Figure 2) and on the medial aspect of the distal thighs and knees. No Gottron papules or nail fold capillary abnormalities were seen. There was symmetrical weakness: 3/5 in the neck flexors, hip flexors, and deltoids and 4/5 in the hands, quadriceps, biceps, and triceps. There was severe tenderness with palpation of the forearms, deltoids, neck, and proximal thigh muscles. Cardiopulmonary examination results were unremarkable.



Figure 1
–
A faint periorbital heliotrope rash and erythematous facial plaques were present in our patient with dermatomyositis. A heliotrope rash is pathognomonic for this condition, although it may be subtle with only mild discoloration.


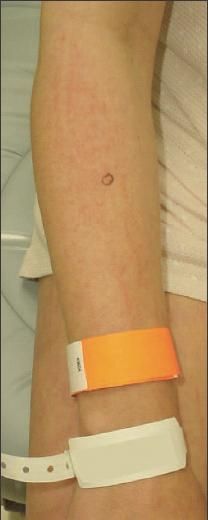
Figure 2 – Some erythematous, linear plaques were present
on the patient's forearms and medial thighs. The patient denied
scratching these areas. The skin lesions of dermatomyositis are
photosensitive; photoprotection measures are an important
component of treatment.
Diagnostic study results were as follows: normal electrolyte levels, complete blood cell count, and thyroid-stimulating hormone levels; C-reactive protein (CRP) level, lower than 0.5 mg/dL; erythrocyte sedimentation rate (ESR), 8 mm/h; AST level, 78 U/L (normal range, 0 to 31 U/L); LDH level, 252 U/L (normal range, 135 to 214 U/L); creatine phosphokinase level, 1730 U/L (normal range, 35 to 150 U/L); aldolase level, 11.8 U/L (normal range, 1.5 to 8.1 U/L); antinuclear antibodies (ANA) titer, lower than 1:40; and electromyography (EMG)/nerve conduction study results consistent with myositis. A skin biopsy revealed interface dermatitis (Figure 3), and a muscle biopsy revealed mild, patchy, lymphocytic perivascular inflammation in the interstitial connective tissue (Figure 4). A myositis-specific antibody panel later came back negative.


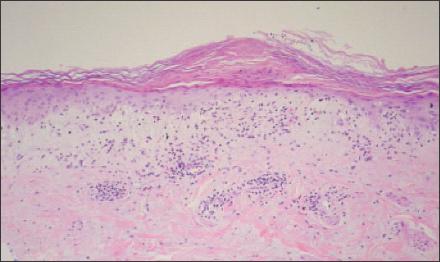
Figure 3
–
Skin biopsy of the patient's right forearm demonstrated lymphocytic inflammation at the dermal-epidermal junction and vacuolar changes at the basal layer. This is consistent with interface dermatitis, which often is found on skin biopsy in patients with dermatomyositis but is nonspecific; it also is present in systemic lupus erythematosus.


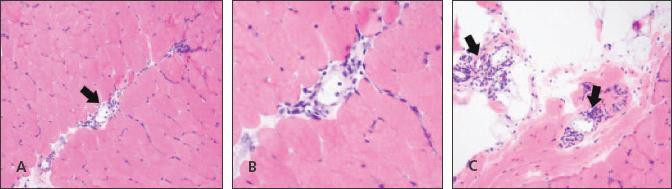
Figure 4
–
A hematoxylin and eosin stained section from the patient's left quadriceps muscle is shown. Mild perivascular lymphocytic inflammation (arrows) is present in the perimysial connective tissue between fascicles (A and B) and in the epimysial fibroadipopse tissue (C).
Despite receiving prednisone, 30 mg twice daily for 2 weeks, the patient had worsening neck weakness and new dysphagia. Fiberoptic endoscopic evaluation revealed pharyngeal weakness. The addition of methylprednisolone, 1g intravenously for 3 days, provided symptomatic improvement starting 4 days after the final methylprednisolone infusion.
The patient subsequently was found to have low IgG levels, and a diagnosis of common variable immunodeficiency was made. Intravenous immunoglobulin therapy-given first at 300 mg/kg and then at 400 mg/kg, every 3 weeks-was beneficial. The patient returned to normal strength, and the rash resolved. Prednisone was tapered to 5 mg/d, and a healthy daughter was delivered. Mother and daughter did well during the first month postpartum.
CLASSIFICATION
For a definitive diagnosis of DM, the Bohan and Peter3,4 classification requires a characteristic rash plus 3 of the following criteria:
• Symmetrical, subacute, proximal muscle weakness.
• Muscle biopsy abnormalities: necrosis, regeneration, fiber size variation, inflammation, and atrophy.
• Muscle enzyme level elevation: CK, aldolase, AST, ALT, and LDH.
• Electromyographic abnormalities: triad of short, small, polyphasic motor units; insertional irritability, positive sharp waves, fibrillations; and bizarre, high-frequency repetitive discharges.
• Rash typical of DM: heliotrope rash or Gottron papules.
Evidence of muscular dystrophy is an exclusion. Others include evidence of neuromuscular, infectious, posttraumatic, toxic, metabolic, and endocrine disease.
DM may occur in association with malignancy, immunodeficiency syndromes,and medication use.5 It may overlap with other connective tissue diseases, especially scleroderma and systemic lupus erythematosus (SLE). Amyopathic DM (ADM) is a special subset that has classic skin findings but no clinical evidence of weakness. About 30% of cases of DM present this way; clinically evident myositis develops in most persons weeks to years later.6
DEMOGRAPHICS
The annual incidence of DM and PM is 2 to 10 cases per million,with a recent trend of increasing incidence7; the prevalence is about 1 case per 100,000. In adult DM, the overall ratio of women to men is 2:1; when overlapping with another connective tissue disease is included, it is 10:1. DM is more common in African Americans. IIMs may occur at any age, but the onset peaks between 10 and 15 years and 45 and 60 years. Only 14% of patients with DM or PM present during childbearing years (age 15 to 30 years).8
CLINICAL FEATURES
A photosensitive rash often is the initial manifestation; it may precede muscle disease by more than a year. The severities of the rash and muscle disease may be parallel or follow disparate courses. Gottron papules (seen in 60% to 80% of patients) and the heliotrope rash (seen in fewer than 50% of patients) are pathognomonic for DM; many other cutaneous lesions are characteristic but not as specific.
Gottron papules are scaly, erythematous plaques symmetrically located on bony prominences, especially the metacarpophalangeal, proximal interphalangeal, and distal interphalangeal joints, although the elbows, knees and feet also may be affected. The heliotrope rash is a purplish discoloration of the upper eyelids, sometimes with periorbital edema. The discoloration may be slight, appearing as only mild discoloration of the eyelid margin (as in our patient's case). Scaling and desquamation may occur in addition to the color changes.
Poikiloderma-a combination of atrophy, dyspigmentation, and telangiectasia-often occurs over the anterior chest ("V sign"), upper back ("shawl sign"), extensor surfaces of the arms, and upper-lateral thighs ("holster sign").9 Nail fold changes include periungual erythema with dilated nail fold capillaries and cuticle overgrowth. Hyperkeratosis and cracking of the lateral and palmar aspects of the fingers are termed "mechanic's hands." Also reported are malar erythema, nonscarring alopecia, psoriaform dermatitis of the scalp, panniculitis, facial seborrhea, and flagellate erythema (linear streaks on the trunk and proximal extremities). Calcinosis of soft tissue is a late complication of juvenile DM, but it is uncommon in adult-onset disease.
Symmetrical, proximal muscle weakness usually is of insidious onset, although acute onset may occur. Distal muscle weakness, such as in the finger flexors, is characteristic of IBM but uncommon in DM and PM. Muscle atrophy may be indicative of IBM, but it is absent in early DM and PM. Unlike in PM, which usually is painless, significant myalgia may occur in DM (as in this patient).
Lower extremity weakness often manifests first, with difficulty in climbing stairs or rising from a chair or toilet. Upper extremity weakness leads to difficulty in combing or washing hair. Neck flexor weakness, evidenced by an inability to lift the head from the pillow, is common. Neck extensor strength remains preserved unless the disease is severe and chronic; weakness suggests another diagnosis (eg, muscular dystrophy or neck extensor myopathy). Pharyngeal weakness manifested by nasal regurgitation, dysphagia, or dysphonia is less common; it portends a poor prognosis. Ocular or facial weakness is rare in DM and PM and suggests other diagnoses.
Constitutional symptoms include fatigue, fever, and weight loss. Fatigue often is severe and persistent even with adequate treatment of other disease features. Fever is especially frequent with antisynthetase syndrome (occurs in a few patients with myositis, who may present with dominant symptoms and signs in tissues other than muscle [fever, mechanic's hands, arthritis, and interstitial lung disease]). Weight loss may be a manifestation of systemic inflammation but also may herald underlying malignancy.
Cardiac and pulmonary involvement could be life-threatening. Pulmonary complications include interstitial lung disease (ILD), aspiration pneumonia, and hypoventilation resulting from respiratory muscle weakness. ILD occurs in 5% to 46% of cases of DM and PM, especially in patients who have antisynthetase antibodies.10 It may be severe and rapidly progressive, especially in ADM,6 resulting in respiratory failure and death. Pharyngeal or esophageal muscle weakness may result in aspiration. Weakness of the diaphragm or intercostal muscles, which is uncommon, may lead to respiratory failure.
Clinically symptomatic cardiac involvement is uncommon. Manifestations include myocarditis (rarely leading to heart failure), pericarditis, valvular disease, and rhythm disturbances. Asymptomatic conduction abnormalities, such as atrial and ventricular arrhythmias, nonspecific ST-T changes, and atrioventricular and bundle-branch blocks, have been observed in up to 72% of patients with DM and PM.11 Polyarthritis,in a rheumatoid arthritis–like distribution, may occur and usually is mild and nonerosive. 7 Raynaud phenomenon, with abnormal nail fold capillaries, is common in DM. Uncommon clinical features include smooth muscle involvement of the intestinal tract, retinopathy with vision loss, proteinuria, and CNS vasculitis.7
Malignancy in DM
Malignancies, predominantly adenocarcinomas, occur in 20% to 25% of DM cases9; they are more common in patients older than 50 years, but they may be seen in any age group. The risk is greatest in the first 3 years after the diagnosis of myositis, but an increased risk persists lifelong. Ovarian cancer is overrepresented in patients with DM, and the diagnosis may be made many years after the onset of the myositis. Increased associations with lung, pancreatic, stomach, colorectal cancer, and non-Hodgkin lymphoma have been reported. Cancer is seen more frequently in patients with cutaneous necrosis of the trunk, leukocytoclastic vasculitis, and ADM.7
DIAGNOSIS AND EVALUATION
DM should be suspected in patients who have proximal muscle weakness and compatible skin findings. A detailed, well-documented strength examination at every visit is important for assessing treatment response.
CK, LDH, AST, ALT, and aldolase all are enzymes present in muscle. The level of at least 1 is elevated in most cases (except for ADM) and often all are elevated. Aldolase and CK are the most sensitive enzymes; however, even their levels are normal in 4% and 5% of cases, respectively. 12 However, myoglobinuria is rare in the IIMs.
The ESR and CRP level are poor markers of disease activity and often are normal or minimally elevated in spite of severe muscle inflammation. ANAs are positive in 50% to 80% of cases. Many of the myositis-specific autoantibodies are directed against cytoplasmic antigens, explaining why ANA test results often are negative. Found in about 20% to 30% of patients with DM or PM, myositis-specific autoantibodies aid in the diagnosis and characterization of the disease and as prognostic indicators of disease severity.13
EMG usually is performed on half of the body, preserving the contralateral side for muscle biopsy. Classic findings in myositis are noted above. Although these findings are not specific for IIM, and may be seen with any cause of myositis, they are an important component of diagnosis-EMG helps rule out neuropathic disorders. The result is abnormal in about 90% of patients with myositis; thus, a normal EMG result is strong evidence against active myositis.12 A muscle injured by needle EMG will have inflammatory histology and should not be biopsied for at least 1 month after the test.2
The muscle biopsy is central to establishing the diagnosis, but selecting the appropriate site for biopsy may be difficult. Muscles with end-stage disease should be avoided because the pathological findings often are nonspecific, showing only atrophy and maybe inflammation. The disease is patchy, and normal muscle may be obtained despite active disease nearby.
Selection of an appropriate muscle for biopsy may be aided by the physical examination (choose a moderately but not severely weak muscle), EMG, and MRI. With short tau inversion recovery images, MRI may detect muscle edema, which may be interpreted as active myositis.14 If possible, preference should be given to biopsy of a standard muscle site (quadriceps or deltoid) that is well characterized in a pathological sense.
DM and PM have overlapping but different pathological findings. Both have lymphocytic inflammation; however, the distribution of the inflammatory infiltrate and the subset of involved lymphocytes differ. In PM, CD8+ T-cells target muscle fibers, resulting in diffuse muscle injury (predominantly endomysial inflammation) without vasculopathy. In DM, B-cells and complement target small blood vessels, causing perivascular inflammation. As a result, there is muscle microinfarction and inflammation around the muscle fascicle (perifascicular inflammation and atrophy).7 Endomysial inflammation is much less frequent in DM.
Skin biopsy often demonstrates inflammatory changes at the dermoepidermal junction (interface dermatitis). This finding is nonspecific and also may be seen in SLE.
Further testing is needed to determine the extent of the disease and evaluate for underlying malignancy. In addition to a thorough history and physical examination, chest x-rays, pulmonary function testing with diffusion capacity, swallow studies, and electrocardiography are important.
An evaluation for cancer, based on the patient's age and sex, is necessary in all cases of DM. The evaluation should include testicular or breast and pelvic examination with Papanicolaou smear. Urinalysis, CA-125 (ovarian cancer is overrepresented in DM), stool occult blood testing, chest x-ray, mammography, and CT of the abdomen and pelvis also should be considered. The American Academy of Dermatology recommends re-evaluation for malignancy every 6 to 12 months for the first 2 years after diagnosis.1
TREATMENT
Physical therapy and occupational therapy should be started at diagnosis. Active disease should not prevent patient-specific graded exercise.14 Exercise is safe and beneficial in reducing disability in patients with IIM.15
Corticosteroids are the foundation of treatment. Oral prednisone usually is initiated at 1 mg/kg/d in divided doses to a maximum of 80 mg/d and continued until the CK level improves (often after 4 weeks). Improvement in muscle strength may lag behind enzyme improvements by 2 or 3 months. In cases with severe manifestations, pulse methylprednisolone, 1000 mg/d for 3 days (as used in this case), may help achieve a more rapid response. Prednisone is then consolidated into a single daily dose and tapered by 20% to 25% every month until 5 to 10 mg/d is achieved.16
Low-dose prednisone usually is continued to complete a year of therapy. Patients should be evaluated for response to therapy monthly, with measurement of muscle enzyme levels, manual muscle testing, and assessment of functional status.
To ameliorate corticosteroid-induced bone loss, the use of calcium, 1500 mg/d (in divided doses with food), and vitamin D, 1000 to 2000 IU/d, is appropriate. Bisphosphonate therapy is indicated when 5 mg/d or more of prednisone is to be used for more than 2 months, but it should be avoided in pregnancy and in women of childbearing age. Chemoprophylaxis against pneumocystis pneumonia also should be initiated.
Most patients require corticosteroid-sparing medication during the course of their disease. Many experts recommend immediate initiation of a corticosteroid-sparing agent concomitant with the start of prednisone, especially in severely ill patients. In one study, 87% of patients with DM responded to corticosteroid therapy initially, but 92% had recurrence with subsequent taper.13
Methotrexate and azathioprine are frequently used first-line corticosteroid- sparing agents. Other agents that have been used include cyclosporine, tacrolimus, cyclophosphamide, rituximab, mycophenolate mofetil, hydroxychloroquine, intravenous immunoglobulin, infliximab, and etanercept.16,17
PROGNOSIS
Most patients with DM have a chronic course with either persistent disease activity or repeated exacerbations and remissions. However, patients with DM are more likely than patients with PM to have a monophasic disease that responds to therapy initially and remains in long-term remission after all therapy is stopped.7
Before the widespread use of corticosteroids, as many as 50% of patients with DM or PM died because of disease complications.7 However, corticosteroid therapy has improved outcomes and survival. In one recent study, 5-year survival for adult patients with DM or PM without associated cancer was 95%.18 A poorer prognosis is associated with older age, malignancy, delayed treatment, pharyngeal dysfunction with aspiration pneumonia, ILD, and clinically evident myocardial involvement.7
Disability accumulates because of the inflammatory muscle disease and complications of corticosteroid and immunosuppressive therapy. Up to one-third of patients will have mild to severe functional disability, and disability increases with disease duration.19
CONCLUSIONS
IIMs are systemic diseases that may be severe and involve more than the shoulder and hip musculature, evidenced by recalcitrant pharyngeal weakness in the patient discussed here. The ability to recognize rash associated with DM is important; as this case illustrates, however, rash may be subtle and require careful examination. IIMs typically are characterized by painless muscle weakness, but DM occasionally presents with striking pain in addition to weakness. Muscle or skin biopsy or both play an important role in confirming the diagnosis.
Practice Points
1. Painless muscle weakness characterizes the idiopathic inflammatory myopathies, but dermatomyositis may include striking pain.
2. The ability to recognize associated rash is important, but rash may be subtle and require careful examination.
3. Muscle or skin biopsy or both play an important role in confirming the diagnosis.
References:
References1. Drake LA, Dinehart SM, Farmer ER, et al. Guidelines of care for dermatomyositis. American Academy of Dermatology. J Am Acad Dermatol. 1996;34:824-829.
2. Dalakas MC. Polymyositis, dermatomyositis and inclusion-body myositis. N Engl J Med. 1991;325:1487-1498.
3. Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292:344-347.
4. Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med. 1975;292:403-407.
5. Dourmishev AL, Dourmishev LA. Dermatomyositis and drugs. Adv Exp Med Biol. 1999;455:187-191.
6. Gerami P, Schope JM, McDonald L, et al. A systematic review of adult-onset clinically amyopathic dermatomyositis (dermatomyositis siné myositis): a missing link within the spectrum of the idiopathic inflammatory myopathies. J Am Acad Dermatol. 2006;54:597-613.
7. Oddis CV, Medsger Jr TA. Inflammatory muscle disease: clinical features. In: Hochberg MC, Silman AJ, Smolen JS, Weinblatt ME, eds. Rheumatology. 3rd ed. Philadelphia: Mosby; 2003:1537-1554.
8. Silva CA, Sultan SM, Isenberg DA. Pregnancy outcome in adult-onset idiopathic inflammatory myopathy. Rheumatology (Oxford). 2003;42:1168-1172.
9. Callen JP, Wortmann RL. Dermatomyositis. Clin Dermatol. 2006;24:363-373.
10. Takada K, Nagasaka K, Miyasaka N. Polymyositis/dermatomyositis and interstitial lung disease: a new therapeutic approach with T-cell–specific immunosuppressants. Autoimmunity. 2005;38:383-392.
11. Lundberg IE. The heart in dermatomyositis and polymyositis. Rheumatology (Oxford). 2006;45(suppl 4):iv18-iv21.
12. Bohan A, Peter JB, Bowman RL, Pearson CM. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore). 1977;56:255-286.
13. Troyanov Y, Targoff IN, Tremblay JL, et al. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients. Medicine (Baltimore). 2005;84:231-249.
14. Coyle KM, Plotz PH, Gourley MF. Why isn't my myositis patient getting better? The Rheumatologist. 2008;2:18, 19, 22.
15. Alexanderson H, Lundberg IE. The role of exercise in the rehabilitation of idiopathic inflammatory myopathies. Curr Opin Rheumatol. 2005;17:164-171.
16. Oddis CV. Idiopathic inflammatory myopathy: management and prognosis. Rheum Dis Clin North Am. 2002;28:979-1001.
17. Quain RD, Werth VP. Management of cutaneous dermatomyositis. Am J Clin Dermatol. 2006;7:341-351.
18. Sultan SM, Ioannou Y, Moss K, Isenberg DA. Outcome in patients with idiopathic inflammatory myositis: morbidity and mortality. Rheumatology (Oxford). 2002;41:22-26.
19. Bronner IM, van der Meulen MF, de Visser M, et al. Long-term outcome in polymyositis and dermatomyositis. Ann Rheum Dis. 2006;65:1456-1461.














