Inflammatory bone loss in joint replacements: The mechanisms
More than 1 million total joint arthroplasties (TJAs) are performed in the United States each year, and much higher demand is projected. Most TJAs are successful, but aseptic loosening of implants remains a problem that limits long-term success. Aseptic osteolysis, an inflammatory reaction, occurs in up to 10% of patients at 10 years postsurgery.
Total joint implants remain one of the miracles of modern medicine, returning mobility and quality of life to millions of persons every year. More than 1 million total joint arthroplasties (TJAs) are performed in the United States each year, and much higher demand is projected with the aging of the baby boomer generation.1,2 Although most TJAs are successful by any outcome measure initially, aseptic loosening of implants remains a problem that limits long-term success. 2,3
I


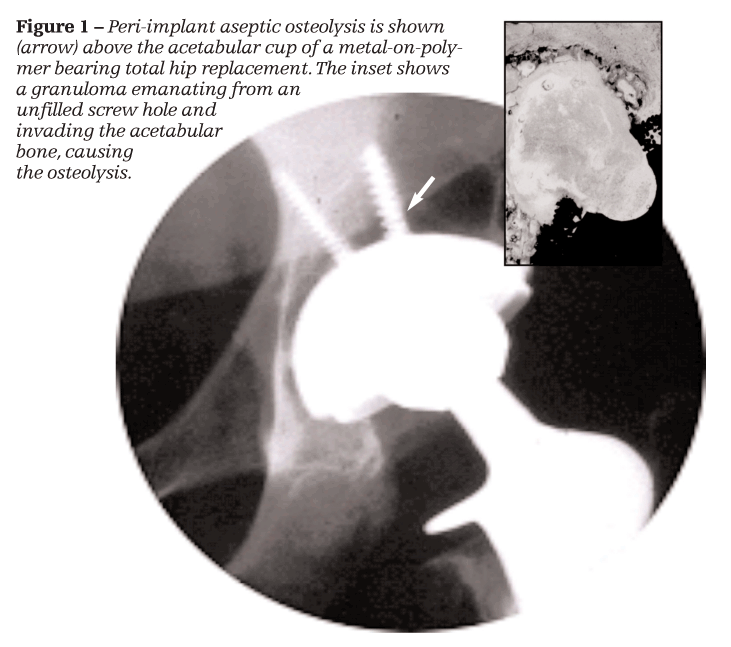
mplant performance varies with the type and amount of degradation produced; all implant debris causes a local inflammatory response that eventually results in bone loss and loss of implant fixation.4 This kind of inflammatory reaction-aseptic osteolysis-occurs in up to 10% of patients at 10 years postsurgery, with marked bone loss around the implant, resulting in pain, premature loosening, and revision.5-7 Aseptic osteolysis generally refers to bone loss around an implant that is observable radiographically (Figure 1).
Knowledge of implant debris–induced inflammation and resulting osteolysis has improved over the years, and advances have been made in understanding the inflammatory bone loss at the molecular and cellular levels. Treatment options, ranging from testing preexisting conditions of metal allergy to general management of inflammation to selective blocking of cellular mediators for hindering of implant debris–induced osteolysis promise to help mitigate the problem of bone loss.
In this 2-part article, we offer current insights into inflammatory bone loss in joint replacements. This first part describes the intracellular mechanisms involved in implant debris–induced osteolysis and new avenues for pharmacological treatment. The second part, to appear in an upcoming issue of this journal, will elaborate on the osteoimmunological science.
Innate immune response
by macrophages
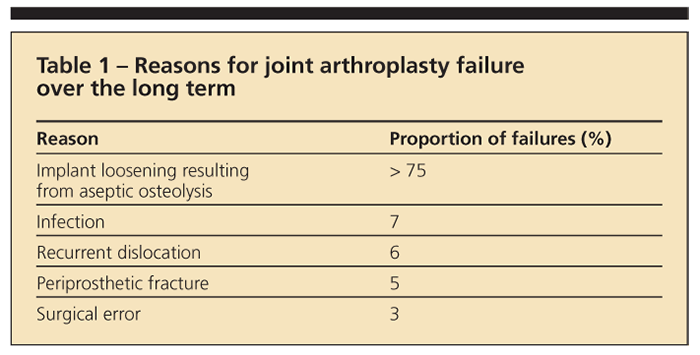
Inflammatory osteolysis has been attributed to an innate immune response by macrophages to implant-generated debris (polymer and metal particles) from implant wear and corrosion. Implant loosening resulting from aseptic osteolysis accounts for more than 75% of joint arthroplasty failures and is the limiting factor in long-term implant performance, but there are additional reasons for long-term implant failure (Table 1).8


Properly positioned implants wear predictably. However, the amount of debris-induced bone loss varies from person to person, even if persons have similar rates of implant wear (implant debris). In fact, some persons with severely worn components may demonstrate little inflammation and periprosthetic bone loss while others with modest amounts of wear may demonstrate extensive osteolysis and implant loosening.
Rates of debris-induced immune reactivity with ensuing aseptic inflammation and subsequent early failure may be as high as 4% to 5% at 6 to 7 years postsurgery in some designs of current-generation metal-on-metal total hip arthroplasties (THAs).9-11 Both particulate and soluble ionic debris induce innate immune system activation, primarily via monocyte or macrophage


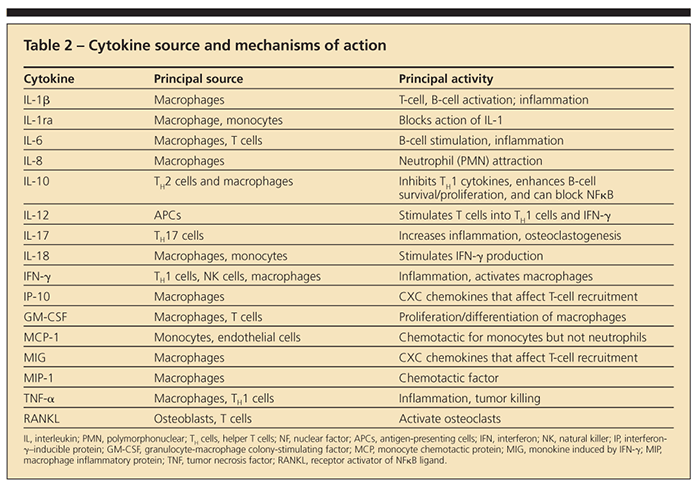
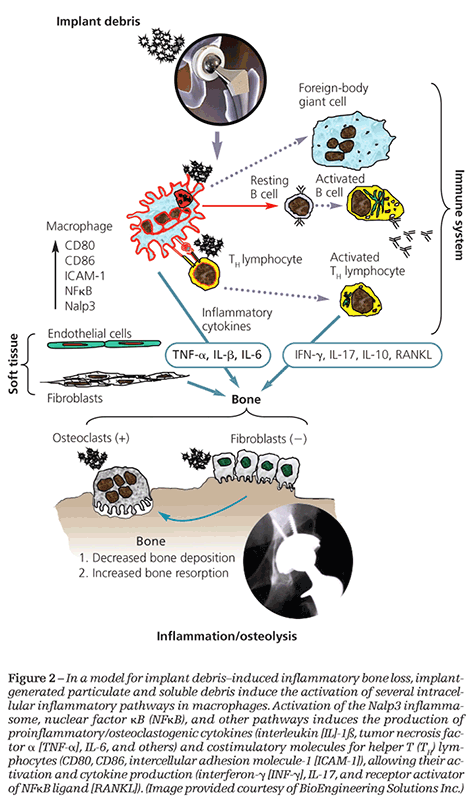
activation,12-18 that up-regulates the transcription factor nuclear factor κB (NFκB) and releases proinflammatory cytokines interleukin (IL)-1b, tumor necrosis factor a (TNF-α), IL-6, αnd IL-8 (Table 2) (Figure 2).12,16,19,20 However, the way that particles and metal ions initiate these proinflammatory responses and which types of implant debris are most reactive is unclear.
The primary goal of the immune system is to protect against harmful pathogens. However, pathogen protection also causes reactivity to implant debris. Both pathogen protection and unwanted reactivity to implant debris are controlled by the sequential activation of innate and adaptive immune systems. The adaptive immune system generally involves production of a very large repertoire of antigen-specific cells; an innate immune response is less antigen-specific, occurs more quickly, and provides the essential signals for adaptive system activation.


Metal sensitivity
Immune reactivity to implant debris has been attributed primarily to innate immune responses, in particular macrophage-induced inflammation. However, a subset of patients experience exuberant adaptive immune system (lymphocyte)–mediated responses to implant debris, in particular, metals.
This so-called metal hypersensitivity to implant materials may be the result of a preexisting condition or may be acquired by having an implant.21 Metal released from implants may form complexes with native proteins that act as antigens. Metals recognized as sensitizers include beryllium, nickel, cobalt, and chromium; occasional responses to tantalum, titanium, and vanadium have been reported. Nickel is the most common metal sensitizer in humans, followed by cobalt and chromium. The temporal and physical evidence associated with the symptoms of type IV allergic responses (including severe dermatitis, urticaria, and vasculitis) to the implantation of orthopedic devices are severe enough to necessitate revision arthroplasty in what is speculated to be about 1% of patients.
Evaluation for metal sensitivity includes skin testing (so-called patch testing or intradermal testing) and in vitro by lymphocyte transformation testing (LTT). Although general patch testing protocols and commercial kits are available for a variety of commonly antigenic substances,22,23 there is continuing concern about sensitizing patients with patch testing24 and about the questionable use of dermal Langerhans cells as accurate diagnostic proxies of peri-implant antigen-presenting cells.25,26 Metal-LTT, or lymphocyte proliferation testing, involves measuring the proliferative response of lymphocytes obtained from peripheral blood by routine blood draw.
Studies that used both of these testing methods found the incidence of metal sensitivity among patients who have metal-containing total joint replacement (TJR) implants to be about 25% (roughly twice as high as that in the general population) and the average incidence of metal sensitivity among patients who have a “failed” implant (in need of revision surgery) to be about 50% to 60%.27-29 These findings indicate that the adaptive immune system may be involved to a much larger degree in debris-induced inflammation than what previously was attributed to the innate immune system via macrophage reactivity.
Causality between implant outcome and level of immune system reactivity to metals has not been well established in the general population. Thus, prescreening all TJA recipients without a history or specific concern about metal allergy is not yet scientifically supported.
Bone remodeling imbalance
Debris-generated inflammatory responses lead directly or indirectly to an imbalance in the rate of bone remodeling (deposition and resorption); osteoclast (bone resorbing cells) activity eventually becomes more prominent, leading to bone loss and loosening of the implant. How does this occur? Macrophages exposed to implant debris particles (of polymer, ceramic, and metal origin) secrete proinflammatory cytokines that activate osteoclast precursor cells as well as mature osteoclasts, aggravating bone destruction and implant loosening (aseptic loosening), with granulomatous tissue invading the bone-implant interface (see Figure 1).
Although understanding of the intracellular mechanisms involved in implant debris–induced osteolysis remains incomplete, much progress has been made in understanding the osteoimmunological science leading to new avenues for pharmacological treatment. Specific intracellular pathways (including particle-induced activation of NFκB and activation of kinase pathways, such as p38, mitogen-activated protein kinases, extracellular signal-regulated kinases, and c-Jun amino-terminal kinases) have been implicated as mediators of implant debris–induced inflammatory cytokines, such as IL-1, TNF-α, and IL-6 (see Figure 2).30,31
Some new pathways, such as the inflammasome danger-signaling pathway, have been identified as possible “danger sensors” that transduce phagocytosis of seemingly benign sterile implant debris into macrophage production of potent cytokines, such as IL-1β.32 However, the immune cell kinetics of implant debris detection and recognition of broad cellular inflammatory response is still incompletely understood.
There is increasing evidence that elevated metal ion concentrations found locally and systemically have the potential to induce toxic and genotoxic effects on peri-implant cells.9,10,33,34 New types of “alternative” bearings, such as metal-on-metal articulating components, have resulted in new types of biological responses, including lymphocyte-dominated granuloma-like lesions in bone (termed “ALVAL,” for aseptic lymphocyte vasculitis–associated lesion) found around some implants. There is evidence of the involvement of the adaptive immune system in tissue samples retrieved from failed arthroplasties where lymphocytic infiltrates have been found.35,36 This highlights the fact that both innate (macrophage) and adaptive (T lymphocyte) immune system reactivity to implant debris can produce inflammatory cytokines that negatively affect bone remodeling and limit the lifetime of current TJR implants.
Pinpointing a mechanism
is difficult
Pinpointing a specific protein, a cell, or even a cellular mechanism as singularly responsible for the complex process of implant debris–induced inflammatory bone loss is difficult. Earlier studies showed excessive quantities of implant-generated particles and metal ion concentrations in periprosthetic tissue retrieved from failed THAs; as much as 109 particles per gram of tissue and up to 2-mM concentrations of cobalt and chromium ions have been detected.9,37-39 In addition to the high concentrations of soluble and particulate wear debris found in the peri-implant environment, tissue retrieved from patients with aseptic loosening also shows sizable infiltrations of immune and other stromal cells, such as macrophages, fibroblasts, and T lymphocytes in the form of granulomas, necrotic lesions, and fibrous tissue.40-42
The evidence of excessive immune reactivity in the periprosthetic tissue is documented by protein analysis, where concentrations of proinflammatory markers (eg, monokine induced by γ-interferon 1, soluble intercellular adhesion molecule 1, TNF-α, IL-1, IL-6, IL-8, and IL-10) are elevated compared with control samples.43 Elevated immune factors triggered by implant debris play an active role in the inflammatory and osteolytic process. In addition, longitudinal studies performed on cohorts of patients who had undergone TJAs, both with measurable periprosthetic osteolysis and without any observable osteolysis, demonstrated a close relationship between the amount of particulate wear debris found in tissue and the degree of bone resorption measured.2,44 These studies concluded that implant wear debris is the major cause of aseptic loosening of implants.
Understanding of the specific mechanisms by which implant wear triggers an inflammatory response remains incomplete. In addition, knowledge about the direct effects of debris-induced inflammation on other peri-implant cells (fibroblasts, osteoblasts, osteoclasts) and how they all contribute to osteolysis and loosening also remains incomplete. This is the subject of current investigation.
Conclusions
Debris produced from implants is minimized but is unavoidable and results in activation of the innate immune system and, sometimes, the adaptive immune system. The result is local inflammation that over time causes bone loss and implant loosening via aseptic osteolysis.
References:
References1. Hallab N. Metal sensitivity in patients with orthopedic implants. J Clin Rheumatol. 2001;7:215-218.
2. Looney RJ, Schwarz EM, Boyd A, O'Keefe RJ. Periprosthetic osteolysis: an immunologist's update. Curr Opin Rheumatol. 2006;18:80-87.
3. Hallab NJ, Anderson S, Stafford T, et al. Lymphocyte responses in patients with total hip arthroplasty. J Orthop Res. 2005;23:384-391.
4. Willert HG, Semlitsch M. Reactions of the articular capsule to wear products of artificial joint prostheses. J Biomed Mater Res. 1977;11:157-164.
5. Arora A, Song Y, Chun L, et al. The role of the TH1 and TH2 immune responses in loosening and osteolysis of cemented total hip replacements. J Biomed Mater Res A. 2003;64:693-697.
6. Jacobs JJ, Roebuck KA, Archibeck M, et al. Osteolysis: basic science. Clin Orthop Relat Res. 2001;393:71-77.
7. Archibeck MJ, Jacobs JJ, Roebuck KA, Glant TT. The basic science of periprosthetic osteolysis. Instr Course Lect. 2001;50:185-195.
8. Holt G, Murnaghan C, Reilly J, Meek RM. The biology of aseptic osteolysis. Clin Orthop Relat Res. 2007;460:240-252.
9. Jacobs JJ, Hallab NJ. Loosening and osteolysis associated with metal-on-metal bearings: a local effect of metal hypersensitivity? J Bone Joint Surg. 2006;88A:1171-1172.
10. Korovessis P, Petsinis G, Repanti M, Repantis T. Metallosis after contemporary metal-on-metal total hip arthroplasty: five to nine-year follow-up. J Bone Joint Surg. 2006;88A:1183-1191.
11. Milosev I, Trebse R, Kovac S, et al. Survivorship and retrieval analysis of Sikomet metal-on-metal total hip replacements at a mean of seven years. J Bone Joint Surg. 2006;88A:1173-1182.
12. Kaufman AM, Alabre CI, Rubash HE, Shanbhag AS. Human macrophage response to UHMWPE, TiAlV, CoCr, and alumina particles: analysis of multiple cytokines using protein arrays. J Biomed Mater Res A. 2008;84:464-474.
13. Hallab NJ, Cunningham BW, Jacobs JJ. Spinal implant debris-induced osteolysis. Spine (Phila Pa 1976). 2003;28:S125-S138.
14. Sethi RK, Neavyn MJ, Rubash HE, Shanbhag AS. Macrophage response to cross-linked and conventional UHMWPE. Biomaterials. 2003;24:2561-2573.
15. Trindade MC, Lind M, Nakashima Y, et al. Interleukin-10 inhibits polymethylmethacrylate particle induced interleukin-6 and tumor necrosis factor-alpha release by human monocyte/macrophages in vitro. Biomaterials. 2001;22:2067-2073.
16. Catelas I, Petit A, Zukor DJ, et al. TNF-alpha secretion and macrophage mortality induced by cobalt and chromium ions in vitro-qualitative analysis of apoptosis. Biomaterials. 2003;24:383-391.
17. Catelas I, Petit A, Marchand R, et al. Cytotoxicity and macrophage cytokine release induced by ceramic and polyethylene particles in vitro. J Bone Joint Surg. 1999;81B:516-521.
18. Campbell PA, Wang M, Amstutz HC, Goodman SB. Positive cytokine production in failed metal-on-metal total hip replacements. Acta Orthop Scand. 2002;73:506-512.
19. Lewis JB, Randol TM, Lockwood PE, Wataha JC. Effect of subtoxic concentrations of metal ions on NFkappaB activation in THP-1 human monocytes. J Biomed Mater Res A. 2003;64:217-224.
20. Lewis JB, Wataha JC, McCloud V, et al. Au(III), Pd(II), Ni(II), and Hg(II) alter NF kappa B signaling in THP1 monocytic cells. J Biomed Mater Res A. 2005;74:474-481.
21. Hallab N, Merritt K, Jacobs JJ. Metal sensitivity in patients with orthopaedic implants. J Bone Joint Surg. 2001;83A:428-436.
22. Hensten-Pettersen A. Allergy and hypersensitivity. In: Morrey BF, ed. Biological, Material, and Mechanical Considerations of Joint Replacement. New York: Raven Press; 1993:353-360.
23. Rooker GD, Wilkinson JD. Metal sensitivity in patients undergoing hip replacement: a prospective study. J Bone Joint Surg. 1980;62B:502-505.
24. Merritt K, Brown SA. Tissue reaction and metal sensitivity: an animal study. Acta Orthop Scand. 1980;51:403-411.
25. Res P, Kapsenberg ML, Bos JD, Stiekema F. The crucial role of human dendritic antigen-presenting cell subsets in nickel-specific T-cell proliferation. J Invest Dermatol. 1987;88:550-554.
26. Friedmann PS. Contact sensitisation and allergic contact dermatitis: immunobiological mechanisms. Toxicol Lett. 2006;162:49-54.
27. Hallab N, Jacobs JJ, Black J. Hypersensitivity associated with metallic biomaterials. In: Wise DL, Trantolo DJ, Lewandrowski KU, et al, eds. Biomaterials Engineering and Devices: Human Applications. Vol 1. Totowa, NJ: Humana Press; 2000:15-24.
28. Hallab NJ, Mikecz K, Vermes C, et al. Differential lymphocyte reactivity to serum-derived metal-protein complexes produced from cobalt-based and titanium-based implant alloy degradation. J Biomed Mater Res. 2001;56:427-436.
29. Hallab NJ, Anderson S, Caicedo M, et al. Immune responses correlate with serum-metal in metal-on-metal hip arthroplasty. J Arthroplasty. 2004;19(8, suppl 3):S88-S93.
30. Beidelschies MA, Huang H, McMullen MR, et al. Stimulation of macrophage TNFalpha production by orthopaedic wear particles requires activation of the ERK1/2/Egr-1 and NF-kappaB pathways but is independent of p38 and JNK. J Cell Physiol. 2008;217:652-666.
31. Wei S, Siegal GP. p38 MAPK as a potential therapeutic target for inflammatory osteolysis. Adv Anat Pathol. 2007;14:42-45.
32. Caicedo MS, Desai R, McAllister K, et al. Soluble and particulate Co-Cr-Mo alloy implant metals activate the inflammasome danger signaling pathway in human macrophages: a novel mechanism for implant debris reactivity. J Orthop Res. 2009;27:847-854.
33. Bitsch RG, Zamorano M, Loidolt T, et al. Ion production and excretion in a patient with a metal-on-metal bearing hip prosthesis: a case report. J Bone Joint Surg. 2007;89A:2758-2763.
34. Merritt K, Rodrigo JJ. Immune response to synthetic materials: sensitization of patients receiving orthopaedic implants. Clin Orthop Relat Res. 1996;326:71-79.
35. Park YS, Moon YW, Lim SJ, et al. Early osteolysis following second-generation metal-on-metal hip replacement. J Bone Joint Surg. 2005;87A:1515-1521.
36. Goodman SB, Huie P, Song Y, et al. Cellular profile and cytokine production at prosthetic interfaces: study of tissues retrieved from revised hip and knee replacements. J Bone Joint Surg. 1998;80B:531-539.
37. Goodman SB, Lind M, Song Y, Smith RL. In vitro, in vivo, and tissue retrieval studies on particulate debris. Clin Orthop Relat Res. 1998;352:25-34.
38. Jacobs JJ, Skipor AK, Campbell PA, et al. Can metal levels be used to monitor metal-on-metal hip arthroplasties? J Arthroplasty. 2004;19(8, suppl 3):S59-S65.
39. Hallab NJ, Caicedo M, Finnegan A, Jacobs JJ. Th1 type lymphocyte reactivity to metals in patients with total hip arthroplasty. J Orthop Surg Res. 2008;3:6.
40. Mahendra G, Pandit H, Kliskey K, et al. Necrotic and inflammatory changes in metal-on-metal resurfacing hip arthroplasties. Acta Orthop. 2009;80:653-659.
41. Kreicbergs A. Pseudotumor after metal fixation of a fracture surgery: a case report. Acta Orthop Scand. 1983;54:739-742.
42. Aroukatos P, Repanti M, Repantis T, et al. Immunologic adverse reaction associated with low-carbide metal-on-metal bearings in total hip arthroplasty. Clin Orthop Relat Res. 2009 Dec 18; [Epub ahead of print].
43. Shanbhag AS, Kaufman AM, Hayata K, Rubash HE. Assessing osteolysis with use of high-throughput protein chips. J Bone Joint Surg. 2007;89A:1081-1089.
44. Wilkinson JM, Hamer AJ, Stockley I, Eastell R. Polyethylene wear rate and osteolysis: critical threshold versus continuous dose-response relationship. J Orthop Res. 2005;23:520-525.














