Strengthening Myositis Decision Making
Patients may have difficulty distinguishing weakness and fatigue, as well as asthenia.
The idiopathic inflammatory myopathies (IIMs), including dermatomyositis (DM), polymyositis (PM), and inclusion body myositis (IBM), are characterized by proximal weakness and inflammation observed in the biopsy specimens of affected muscles (myopathy is an abnormal muscle condition; myositis is muscle inflammation). The main symptoms-weakness, fatigue, and muscle pain-are common complaints in primary care medicine that involve a diverse differential diagnosis with extensive overlap.
DM and PM are autoimmune diseases, and the symptoms often overlap with those of other autoimmune diseases, such as scleroderma, rheumatoid arthritis (RA), and systemic lupus erythematosus (SLE). In addition, muscle inflammation may be seen on muscle biopsy specimens with viral, bacterial, and parasitic infections; sarcoidosis; eosinophilic myositis and macrophagic myofasciitis; and some muscular dystrophies.
Accurate diagnosis is essential for early and aggressive initiation of immunosuppressive therapy. In most cases, a physician may distinguish among the myopathies with a careful history and physical examination, combined with appropriate diagnostic testing. Once a diagnosis has been made, however, numerous issues arise about the disease and the medications used in treatment. Many complex issues may become active simultaneously, creating the risk that less active issues are neglected.
Close collaboration between the primary care physician and the physician who is managing the IIM is critical to the patient's overall care and well-being. In this article, I discuss the issues that often arise in IIM diagnosis and management and how physicians can address them with improved interaction with colleagues and patients.
PATIENT PRESENTATION
Clinically, a patient with an IIM almost always presents with muscle weakness, usually with elevated muscle enzyme levels (eg, creatine phosphokinase [CPK]) and sometimes with myalgia (muscle pain). However, most patients with these complaints probably do not have an IIM because many patients who have other entities present with one or all of the symptoms; IIM is rare (incidence, about 1 case per 100,000 persons),1 and millions of patients are now taking lipid-lowering agents that may cause a toxic myopathy.2 Distinguishing DM and PM from other forms of myopathy is important, however, because these IIMs can be managed successfully with aggressive immunosuppressive therapy.
APPROACH TO PATIENTS WITH WEAKNESS OR FATIGUE
Is the patient weak?
Patients may have difficulty distinguishing weakness and fatigue, as well as asthenia (a feeling of exhaustion or a lack of energy in the absence of muscle weakness).3,4 Asthenia may be associated with excessive sleepiness, difficulty in concentrating, and depression; many patients refer to it as fatigue, but a decreased ability of a muscle to perform repetitive tasks is fatigability. In contrast, weakness is a reduction in the power of a muscle (eg, decreased ability to perform a task on the first try).
These 3 features may be confused easily, especially because they may occur in combination. For example, asthenia is a common feature of many autoimmune conditions (eg, RA and scleroderma) that is thought to be a consequence of the patient's inflammatory burden. If a patient with an autoimmune condition complains of increased weakness or fatigue, this complaint could represent a flare of the underlying illness, an evolution into an inflammatory myopathy, an adverse effect of a medication (eg, corticosteroids causing myopathy), or worsening depression associated with the chronic illness.
Clues in the history that may suggest weakness or fatigability rather than asthenia include difficulty in climbing stairs without using a banister, rising from a chair or toilet without using one's arms, and stepping up onto a curb or onto a bus.The presence of pain, whether resulting from the underlying disease or from a separate process (eg, difficulty in climbing stairs in a patient with knee osteoarthritis) may confound the assessment of strength.
When is the patient weak?
Episodic weakness or weakness after activity has a different implication than does persistent weakness. Depending on the muscle group affected, episodic weakness may suggest transient ischemic attacks, myasthenia gravis, periodic paralysis syndromes, electrolyte abnormalities, or metabolic myopathies. However, most types of myopathy cause persistent weakness.
Where is the patient weak?
The physical examination may be used to confirm suspected weakness and determine the pattern of weakness. The pattern may provide clues about the underlying disorder.
Testing
Manual muscle testing (MMT) is the most common method of strength assessment. The examiner positions a muscle group and exerts a force opposite to the force generated by the muscle group tested. The specific method used may vary considerably among examiners.
To start, the examiner should consider what would be normal strength for each patient. For example, a 75-year-old, 109-lb woman would be expected to be weaker than a 38-year-old, 150-lb man. In addition, the height and strength of the examiner affects his or her interpretation of the patient's strength. Therefore, each examiner should develop a consistent method for assessing strength.
There are several grading systems. The British Medical Research Council system, which is used frequently, has the following 6 grades: 5, normal; 4, movement against gravity and resistance to force from the examiner (examiners often add a minus or a plus to indicate resistance to strong, moderate, or weak applied forces); 3, active movement against gravity only; 2, active movement in a position only with gravity eliminated; 1, trace contraction; and 0, no contraction.5 Note that the system does not imply that a person with a grade of 4 or 3 is 20% or 40% weaker than normal, respectively.
When a patient's strength is being tested, one of the examiner's hands should be placed just proximal to the joint of the target muscle group to stabilize the joint; the hand that will exert force should be placed just proximal to the next distal joint of the target muscle group. Bilateral proximal and distal muscle groups should be tested (eg, deltoids, biceps, wrist extensors, psoas, quadriceps, tibialis anterior, and neck flexors). Known joint instability, damage, or pain should be considered in the choice of muscle groups to be tested. Of note, weakness in facial muscles may suggest specific types of neuromuscular disorders. Other confounding factors include pain, asthenia, comprehension, and the patient's motivation and emotional status.
Testing for sensory loss and reflex testing also should be performed because these factors usually are spared in patients with myopathy. A normal MMT result in a patient complaining of weakness or fatigue may suggest asthenia, but if the patient is much stronger than the examiner, appreciating the reasons why the patient feels he is weak is important.
Weakness may be caused by disorders of upper motor neurons, lower motor neurons, the neuromuscular junction, or the muscles themselves; the pattern of weakness may provide a clue to which one is affected.6 Abnormalities of upper motor neurons may be suggested by the presence of hyperactive reflexes, a positive Babinski sign, distal greater than proximal weakness, or spasticity.
Weakness on one side of the body (hemiparesis) is highly suggestive of upper motor neuron disease (eg, ischemic stroke, intracranial bleed, or brain or spinal cord masses). However, upper motor neuron disease also may cause weakness in both arms, both legs, all 4 extremities, or 1 extremity. In these cases, the presence of upper motor neuron signs, language or cognitive abnormalities, or cranial nerve abnormalities may suggest upper motor neuron disease.
Abnormalities of lower motor neurons may be suggested by the presence of hypoactive reflexes, muscle twitching (fasciculations), distal greater than proximal weakness, or rapid muscle atrophy. Weakness in only one extremity usually is caused by lower motor neuron disease (eg, an abnormality associated with a nerve root, peripheral nerve, or nerve plexus). In these cases, the weakness may be accompanied by neuropathic pain or numbness or both. However, lower motor neuron disease also may cause weakness in both arms, both legs, or all 4 extremities; in these cases, the presence of lower motor neuron signs may help distinguish the etiology.
Myopathy often causes persistent symmetrical proximal muscle weakness with sparing of the facial muscles, but there are many exceptions. Intermittent weakness may suggest a disease of the neuromuscular junction, such as myasthenia gravis, especially if the patient has facial involvement. Involvement of the face and scapular winging suggests facioscapulohumeral dystrophy.
Additional studies
Initial laboratory workup for suspected myopathy should include thyroid function tests; serum muscle enzyme levels (as indirect evidence of muscle breakdown), such as creatine kinase (CK), aldolase, aspartate aminotransferase/alanine aminotransferase (AST/ALT), and lactate dehydrogenase; electrolytes; complete blood cell (CBC) count (an elevated white blood cell count may suggest infection; anemia may suggest inflammatory disease); erythrocyte sedimentation rate (ESR) or C-reactive protein (CRP) level (both are nonspecific markers of inflammation; high levels could suggest infection, systemic inflammatory disease, or underlying malignancy); and, if inflammatory myopathy is suspected, antinuclear antibody panel. CK is the most specific of the muscle enzymes, and the level often is used as an indirect marker of muscle injury.
Although the AST/ALT levels are used frequently to assess liver function, remembering that AST/ALT also may be released from injured muscle is important; such a release may lead to a mistaken suspicion of hepatic tissue injury. Measuring the γ-glutamyl transferase level may help the examiner avoid performing an unnecessary liver biopsy in a patient with skeletal muscle disease. This point is of particular importance with patients who are receiving known hepatotoxic drugs (eg, methotrexate [MTX]), especially if they have a known systemic inflammatory illness (eg, RA or SLE) that can evolve into an inflammatory myopathy. The serum myoglobin level also is specific for muscle injury.
Electromyography may help examiners differentiate neuropathic from myopathic causes of weakness in cases in which the etiology is unclear. The test involves placing needle electrodes in various muscle groups and measuring the muscle potentials, both at rest and during motion. Myopathies cause characteristic abnormal potentials, but distinguishing among myopathies often is not possible. Because the results are operator-dependent, referring the patient to someone experienced with the technique is important. Because the test can be quite painful, warn the patient.
DIFFERENTIAL DIAGNOSIS OF MYOPATHY
The differential diagnosis of true muscle weakness includes endocrine and electrolyte abnormalities, infections, and the effects of toxins/drugs (Table).7,8 The signs and symptoms of DM and PM have significant overlap with other myopathies,9 and the presence of inflammation within muscles examined for histology is not a specific finding.10 As a result, many noninflammatory myopathies may satisfy the classic criteria described by Bohan and Peter11 that typically are used in diagnosis of DM and PM. This can lead to instances in which patients who have noninflammatory myopathy are treated with immunosuppressives or those with inflammatory myopathy are not treated. Several entities may be confused with IIM.


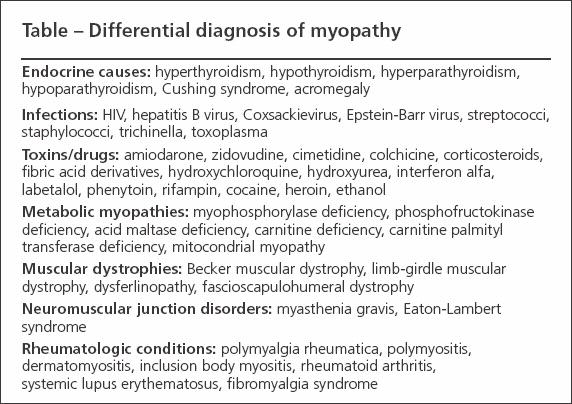
Table
Endocrine causes
Hypothyroidism and hyperthyroidism, hypoparathyroidism and hyperparathyroidism, acromegaly, and Cushing syndrome all may be associated with proximal muscle weakness; however, hypothyroidism is the most likely to cause an elevated CPK level. A diagnosis of hypothyroidism, hyperthyroidism, hypoparathyroidism, or hyperparathyroidism may be made by testing thyroid hormone, parathyroid hormone, calcium, and phosphorus levels.
Infections
Viruses, bacteria, and parasites all may cause muscle inflammation. Viruses may induce diffuse weakness, myalgia, or an elevated CPK level. Those reported to do so include HIV, Coxsackie, rubella, Epstein-Barr, and hepatitis B.
The illnesses usually are self-limited and associated with other signs and symptoms of infection. Bacterial infections of muscle often are caused by staphylococci or streptococci, usually are focal, and may be associated with abscess formation (pyomyositis).
Toxic myopathies
The lipid-lowering agents are a frequent cause of toxic myopathy. However, many other drugs and toxins may cause the condition.12,13
In most cases, stopping the offending agent leads to a reversal of symptoms, although the duration of symptoms after withdrawal is variable and may persist for months. Therefore, taking a detailed history of medication, alcohol, and recreational drug use is important.
Because of the widespread use of lipid-lowering drugs, statin myopathy, myopathy secondary to fibric acid derivatives, or a combination of them is a frequently encountered clinical problem in primary care.2,14,15 Symptoms that may occur in isolation or combination include myalgia, an elevated CPK level, weakness (usually proximal but may be distal) and, rarely, rhabdomyolysis.The myopathic effects of the statin drugs appear to be dose-dependent; vigorous exercise may exacerbate the symptoms.
Baseline CPK screening is recommended for patients who have myopathy symptoms at baseline, those with renal or hepatic dysfunction, and those taking medications known to interact with statins. During therapy, monitoring of the CPK level is recommended only for patients who present with muscle symptoms. Severe symptoms should prompt discontinuation of the drug; mild symptoms may necessitate close monitoring or lowering the dose.
Metabolic myopathies
This is a large, heterogeneous group of disorders that are similar in that they affect the ability of muscle to use energy.16 Patients with a metabolic myopathy have defects in the pathway of carbohydrate breakdown (the muscle glycogenoses), in lipid metabolism, or in mitochondrial DNA. In most cases, the onset is in childhood or in the second through fourth decade of life.
The symptoms may be indistinguishable from those of an IIM (eg, progressive, persistent proximal muscle weakness) but often manifest as episodic fatigue, muscle aches, or cramps after activity; the CPK level may be elevated. Rhabdomyolysis may occur after strenuous activity. Muscle glycogenoses include myophosphorylase deficiency (McArdle disease) and phosphofructokinase deficiency.
Because most of these entities are inherited, patients often have a family history of muscle weakness or fatigability after exercise; therefore, a careful family history can help in the diagnosis. The forearm ischemic exercise test may be used to screen for some of the inborn errors of glycogen metabolism but often yields false-positive results.16 MRI can help differentiate the metabolic myopathies from the IIMs, but inflammation usually is not observed on MRI of the involved muscle group. Muscle biopsy with analysis of the tissue using histochemistry, biochemistry, or electron microscopy is the most useful for making the diagnosis.
Muscular dystrophies
This is a large group of genetic disorders that result in mutation in the genes of structural proteins within a complex of sarcolemmal proteins and glycoproteins.17 Dystrophin and dysferlin are included in this protein complex, but if the genes are mutated, many other associated proteins and glycoproteins can cause disease.
Although the clinical presentation of muscular dystrophies may be indistinguishable from that of IIM, several features may help distinguish them from inflammatory myopathy.These features include a tendency to present in childhood or adolescence, a slowly progressive course, and early muscle atrophy. Muscle biopsy with abnormal staining for the dystrophin complex may help distinguish the muscular dystrophies from other forms of myopathy.
Other disorders
Because patients who have IIM may present with muscle pain or tenderness, fibromyalgia syndrome (FMS) and polymyalgia rheumatica (PMR) may be confused with IIM and these characteristic symptoms may confound the ability to perform accurate MMT. There is no inflammation and no muscle injury in FMS; therefore, the ESR, CRP level, and muscle enzyme levels should be normal. The ESR and CRP level usually are elevated in PMR.
Diseases of the neuromuscular junction include myasthenia gravis and Eaton-Lambert syndrome. They are the result of production of autoantibodies that interfere with critical proteins involved with transmission of signal at this junction. They may present as symmetrical proximal muscle weakness, but weakness usually worsens with repetitive muscle use, facial muscles may be involved, and muscle enzyme levels usually are normal.
CLINICAL AND PATHOLOGICAL FEATURES
DM and PM present with progressive weakness (worsening over weeks to months). The weakness usually is symmetrical and proximal in all extremities, causing difficulty with activities of daily living. If distal muscles become involved, usually it is late in the course. Patients with IBM will more likely have a slower progression of weakness, asymmetrical muscle involvement, and early involvement of distal muscle groups.
Pulmonary involvement, common in the IIMs, may include aspiration pneumonia (as a result of dysphagia) or hypoventilation resulting from weakness of the respiratory muscles. Pulmonary function testing should be performed in patients presenting with acute IIM.
In addition, bronchiolitis obliterans with organizing pneumonia, nonspecific interstitial pneumonia, and interstitial lung disease (ILD) may be seen as a consequence of the autoimmune attack on the lung. ILD occurs in about 10% to 50% of patients with an inflammatory myopathy and is more frequent in patients with autoantibodies to the tRNA synthetases.18
Dermatomyositis
DM affects all age groups (female to male ratio, 3:1). A characteristic rash may appear before the onset of weakness. In some cases, rash may be present with little or no evidence of myopathy (amyopathic or hypomyopathic DM). Characteristic skin manifestations include violet discoloration in a periorbital location or on the extremities (heliotrope or lilac rash; in the periorbital region, swelling usually is present); Gottron papules (erythematous, raised areas over the bony prominences of the hands, elbows, and knees); macular erythema in a V-shape on the chest (V-sign) and over the posterior thorax (shawl sign); and mechanic's hands (cracked areas of skin on the palms and radial aspects of the fingers).
With routine histological examination, a physician may observe cells of the monocyte/macrophage lineage and CD4+ T lymphocytes forming infiltrates arranged in a perivascular distribution in the superficial dermis. The immunohistochemistry of the skin biopsy specimen shows immune complex deposition and evidence of complement activation products at the dermoepidermal junction.
The muscle biopsy results of patients with DM may help differentiate this condition from the other inflammatory myopathies. Atrophic muscle fibers may be observed at the periphery of the fascicle. In addition, cellular infiltrates composed of B lymphocytes and CD4+ T lymphocytes are found in both a perifascicular and perivascular distribution. Complement components and immunoglobulins line the capillary walls, as seen by immunohistochemistry.19
Polymyositis
Although the clinical features of PM are similar to those of DM (except for the rash), the histological findings are distinct. In the muscle biopsy specimens of patients with PM, cellular infiltrates composed mainly of CD8+ T lymphocytes may be observed surrounding and invading normal muscle fibers. By immunohistochemistry, myofibers may be seen to express major histocompatibility complex class 1 on their surface.19 The T-cell receptor repertoire observed in lymphocytes that have been isolated from the muscles of these patients shows restriction when compared with the lymphocytes of the peripheral circulation; this suggests an antigen-driven, oligoclonal T-cell expansion.20
Inclusion body myositis
This is the most common type of myopathy in patients older than 50 years.The patient's weakness progresses slowly; diagnosis often is delayed by months to years.
IBM may be differentiated from the other IIMs clinically by the pattern of muscle involvement; the pattern becomes more evident as the disease progresses.There may be asymmetrical involvement of muscle groups with early distal weakness affecting the wrist and finger flexors. Sensory neuropathy also may be present, a finding not usually seen in DM or PM. In addition, patients with IBM typically respond poorly to therapy.
Findings on muscle biopsy may show the presence of vacuolated muscle fibers with basophilic granular deposits, varying degrees of cellular infiltrate, and intrafiber protein aggregates. If these findings are not present on the specimen sampled for biopsy, distinguishing IBM from PM may be difficult.
The protein aggregates observed have similarities to those found in Alzheimer disease; they consist of β amyloid or phosphorylated Ï proteins.21 Other proteins involved in the processing and folding of amyloid precursor protein also have been found to be associated with these intracellular inclusions. This may suggest that abnormalities in protein handling may cause large amounts of misfolded protein to accumulate within and cause damage to muscle fibers; the damage may trigger inflammation.22
CONSIDERATIONS AFTER THE DIAGNOSIS
Physicians usually prescribe high dosages of corticosteroids (eg, prednisone, 60 mg/d) for patients who are being treated for inflammatory myopathy; although the dosage will be decreased, therapy usually is continued for months to years. In addition, they often prescribe an immunosuppressive agent, such as MTX, azathioprine (AZA), mycofenolate mofetil (MMF), or intravenous immunoglobulin. These medications work much more slowly than corticosteroids, but when they do take effect, they may allow for a reduction of the corticosteroid dosage.
Although corticosteroids usually work well to control the inflammatory myopathy, the use of high dosages for a prolonged period causes numerous adverse effects. Patients with diabetes mellitus (DM) probably will see a worsening of their glycemic control and may even require insulin. New-onset corticosteroid-induced DM may develop in others; it may not resolve as the corticosteroid dosage is reduced.
Hypertension may occur, or there may be a worsening of preexisting hypertension that requires initiation or adjustment of hypertensive agents. Other common adverse effects of corticosteroid use include weight gain, thinning of the skin, easy bruising, cataracts, glaucoma, insomnia, mood changes, difficulty in concentrating, and muscle cramping.
Loss of bone density resulting from corticosteroid use may begin within weeks of taking the first dose. Therefore, patients should begin calcium and vitamin D supplementation immediately, and the use of a bisphosphonate should be considered. In patients who are taking corticosteroids long-term, bone density testing should be performed every year.
Corticosteroid myopathy is a toxic myopathy that can occur when patients take moderate to high doses of corticosteroids. It does not usually cause an elevation of the CPK level, but it can complicate the clinical picture. For example, if a patient with an IIM who has responded to corticosteroids suddenly becomes weaker, whether the weakness is the result of a flare of illness or of corticosteroid myopathy may not be clear. Muscle enzyme level testing and MRI may help the examiner make the distinction. Corticosteroid myopathy usually resolves as the corticosteroids are tapered.
MTX, AZA, and MMF interfere with differentiation and proliferation of cells critical to the adaptive immune response and, therefore, the autoimmune disease. Typical adverse effects with their use include leukopenia and hepatotoxicity. Therefore, the CBC count and liver function test results should be monitored every 1 to 2 months once the patient is on a stable dosage and more frequently when these agents are started.
Patients taking these drugs (and corticosteroids) may be more susceptible to infection than other patients, and the typical signs and symptoms of infection may be masked by these medications. Therefore, the physician should have a high index of suspicion for infection in these patients.
There is an increased incidence of malignancy in patients with DM and PM.23 Cancers often occur 1 to 3 years after the diagnosis of IIM. Suggestions for malignancy screening include gynecological evaluation; mammography; fecal occult blood testing or colonoscopy; prostate-specific antigen testing; the CA-125 test; and CT of the chest, abdomen, and pelvis. Curative management of the cancer may lead to remission of the myositis.
References:
References
- Oddis CV, Medsger TA. Inflammatory muscle disease: clinical features. In: Hochberg MC, Silman AJ, Smolen JS, et al, eds. Rheumatology. 3rd ed. St Louis: Mosby; 2003:1537-1545.
- Baer AN, Wortmann RL. Myotoxicity associated with lipid-lowering drugs. Curr Opin Rheumatol. 2007;19:67-73.
- Mendell JR. Approach to patients with muscle disease. In: Kasper DL, Fauci AS, Longo DL, et al, eds. Harrison’s Principles of Internal Medicine. 16th ed. New York: McGraw-Hill; 2005.
- Saguil A. Evaluation of the patient with muscle weakness. Am Fam Physician. 2005;71:1327-1336.
- Banwell BL, Gomez MR. The clinical examination. In: Engel AG, Franzini-Armstrong C, eds. Myology: Basic and Clinical. 3rd ed. New York: McGraw-Hill; 2004:599-618.
- Olney RK. Weakness, disorders of movement, and imbalance. In: Kasper DL, Fauci AS, Longo DL, et al, eds. Harrison’s Principles of Internal Medicine. 16th ed. New York: McGraw-Hill; 2005.
- Baer AN. Differential diagnosis of idiopathic inflammatory myopathies. Curr Rheumatol Rep. 2006;8:178-187.
- Nirmalananthan N, Holton JL, Hanna MG. Is it really myositis? A consideration of the differential diagnosis. Curr Opin Rheumatol. 2004;16:684-691.
- Amato AA, Griggs RC. Unicorns, dragons, polymyositis, and other mythological beasts.Neurology. 2003;61:288-289.
- Nagaraju K, Plotz PH, Miller FW. Inflammatory muscle disease: etiology and pathogenesis. In: Hochberg MC, Silman AJ, Smolen JS, et al, eds. Rheumatology. 3rd ed. St Louis: Mosby; 2003:1523-1536.
- Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med. 1975;292:403-407.
- Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292:344-347.
- Walsh RJ, Amato AA. Toxic myopathies. Neurol Clin. 2005;23:397-428.
- Christopher-Stine L. Statin myopathy: an update. Curr Opin Rheumatol. 2006;18:647-653.
- Harper CR, Jacobson TA. The broad spectrum of statin myopathy: from myalgia to rhabdomyolysis. Curr Opin Lipidol. 2007;18:401-408.
- Wortmann RL, DiMauro S. Differentiating idiopathic inflammatory myopathies from metabolic myopathies. Rheum Dis Clin North Am.2002;28:759-778.
- Brown RH, Mendell JR. Muscular dystrophies and other muscle diseases. In: Kasper DL, Fauci AS, Longo DL, et al, eds. Harrison’s Principles of Internal Medicine. 16th ed. New York: McGraw-Hill; 2005.
- Schnabel A, Reuter M, Biederer J, et al. Interstitial lung disease in polymyositis and dermatomyositis: clinical course and response to treatment. Semin Arthritis Rheum. 2003;32:273-284.
- Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362:971-982.
- Chinoy H, Ollier WE, Cooper RG. Have recent immunogenetic investigations increased our understanding of disease mechanisms in the idiopathic inflammatory myopathies? Curr Opin Rheumatol. 2004;16:707-713.
- Fratta P, Engel WK, Van Leeuwen FW, et al. Mutant ubiquitin UBB+1 is accumulated in sporadic inclusion-body myositis muscle fibers. Neurology. 2004;63:1114-1117.
- Askanas V, Engel WK. Proposed pathogenetic cascade of inclusion-body myositis: importance of amyloid-beta, misfolded proteins, predisposing genes, and aging. Curr Opin Rheumatol. 2003;15:737-744.
- Levine SM. Cancer and myositis: new insights into an old association. Curr Opin Rheumatol. 2006;18:620-624.
















