Taking a Practical Approach to Giant Cell Arteritis
Laboratory findings, diagnostic methods, and best treatments for giant cell arteritis, a major cause of vision loss and other health problems.
ABSTRACT: Giant cell arteritis (GCA) is a major cause of vision loss and other health problems. Classic histopathological findings include transmural inflammation with mononuclear cells and multinucleated giant cells. Laboratory investigation often demonstrates elevated acute phase reactant levels. GCA symptoms develop abruptly in some patients but more often occur insidiously. Symptoms include headache, scalp tenderness, jaw claudication, and shoulder and hip pain and stiffness. Polymyalgia rheumatica occurs in up to half of patients with GCA. Some patients have aortic involvement. Information from both temporal artery biopsies and MRI studies can assist in making the diagnosis. High-dose corticosteroids and low-dose aspirin are the cornerstones of therapy, but corticosteroids have numerous adverse effects. (J Musculoskel Med. 2008;25:20-28)
Giant cell arteritis (GCA) is the most common form of primary systemic vasculitis. The disease preferentially affects the extracranial branches of the carotid arteries and, less often, causes clinical involvement of the aorta and its major branches.
GCA has an annual incidence of 20 cases per 100,000 persons older than 50 years; in the United States, the prevalence is estimated at 200 cases per 100,000 persons.1 The disorder occurs almost exclusively in persons older than 50 years, exhibits a 3:1 female-to-male predominance, and has a predilection for persons of northern European ancestry.
In patients with GCA, symptoms develop abruptly in a minority of patients but more typically occur in an insidious fashion and remain undiagnosed for weeks or months. The diagnosis is a clinical one, aided by information from temporal artery biopsies and, in some cases, MRI studies of the aorta and its primary branches. Classic histopathological findings include transmural inflammation with mononuclear cells and multinucleated giant cells. Laboratory investigation often demonstrates elevated acute phase reactant levels.
If not recognized promptly and managed appropriately, GCA may be associated with significant morbidity and mortality. High-dose corticosteroids are a cornerstone of therapy, but long-term management of this disorder may be challenging because of their numerous adverse effects.
In this article, we describe a practical approach to making a diagnosis of GCA. We also offer guidelines for treatment with corticosteroids and other agents.
DIAGNOSIS
Clinical manifestations
The clinical presentation of GCA varies from patient to patient, but the classic symptoms and signs are well recognized. Most patients present with indolent symptoms that often are mistaken for infections or malignancies. The textbook symptoms include headache, scalp tenderness, jaw claudication, pain and stiffness in the shoulder and hip areas, weight loss, low-grade fever, and a general sense of being unwell.
In some cases-about 10%-the event that brings patients to medical attention is vision loss. This often occurs against a background of other symptoms that have gone unrecognized by practitioners or even by patients.
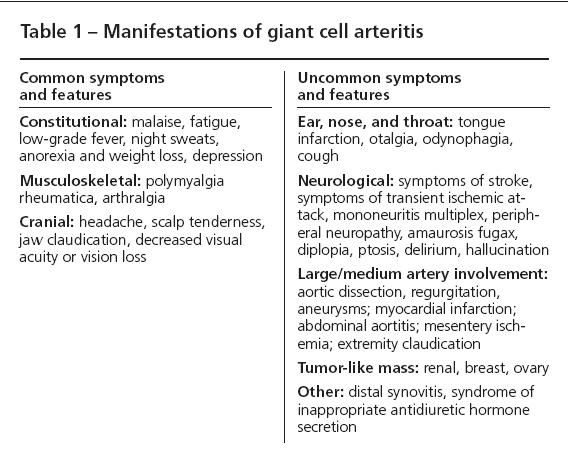
In some patients, cerebrovascular involvement caused by carotid or posterior circulation involvement results in strokes. However, true intracerebral vasculitis in GCA is extremely rare. Several atypical presentations are listed in Table 1, along with more characteristic presentations.


Headaches, often the first disease manifestation, vary in location and character. They may be dull, burning, or throbbing and have a range of severity from patient to patient. The main feature is that the headaches are new for that particular patient. Patients often complain of scalp tenderness and may have difficulty in washing or combing their hair.
Jaw claudication-pain in the masseter muscles caused by chewing-comes on surprisingly quickly after the start of chewing and improves with rest. Because patients do not complain of “jaw claudication,” the physician must elicit this symptom by taking a careful history and searching for synonyms.
Polymyalgia rheumatica (PMR), a syndrome of musculoskeletal pain and stiffness in the neck, shoulder, and hip girdle region, occurs in one third to one half of patients with GCA.1 Patients with PMR often note profound morning stiffness and find it difficult to raise their arms above their head or rise from a chair because of pain. Pain must be distinguished from true weakness as a cause of these disabilities. A few patients with GCA (about 20%) and PMR have a distal synovitis that may be difficult to differentiate from rheumatoid arthritis.
A significant subset of patients with GCA have clinically evident involvement of the aorta and its major branches, eg, the subclavian or carotid arteries. The true frequency of aortic disease in GCA probably is underestimated because to date, no studies have evaluated patients extensively and systematically with imaging protocols (eg, MRI) designed to assess large-vessel involvement. However, aortic involvement has been described in up to 16% of patients (identified in a variety of ways) in a population-based study.2 In most cases, clinically evident large-vessel disease did not emerge until several years after the original diagnosis.
Aortitis and other large-vessel disease in GCA may result in aortic dilatation, aneurysm formation, aortic valve insufficiency, the finding of pulselessness in the upper extremities (because of subclavian and axillary artery disease), and other complications.3 Patients who have GCA need to be monitored carefully for large-vessel involvement with physical examinations and periodic imaging studies (eg, chest radiographs or MRI angiograms).
Physical examination
In some patients, the physical examination reveals features that are highly suggestive of GCA. The absence or diminution of pulses at the brachial or radial sites suggests proximal large-vessel disease. Bruits may be detected in the carotid, subclavian, abdominal aortic, and femoral arteries. Abdominal aortic aneurysms are detected in a few patients, particularly those who have long-standing disease.
Palpation of the temporal arteries may show asymmetrical, diminished, or absent pulses in 1 or both vessels, with associated scalp and temporal area tenderness. Cardiac auscultation can detect diastolic murmurs that suggest aortic insufficiency or dissection. Difficulty with ambulation or with climbing onto the examining table because of pain may suggest PMR.
The most common syndrome of vision loss in GCA is anterior ischemic optic neuropathy (AION), which is caused by progressive stenosis of the ophthalmic or posterior ciliary arteries, leading over time to vascular occlusion. If vision loss through AION has occurred, an afferent papillary defect will be present. If the event causing AION is more than 24 hours old, ophthalmoscopy will reveal blurring of the optic disk. Cotton wool spots and other indications of ocular ischemia also may be present, even before vision loss. Other less common mechanisms of vision loss in GCA (posterior ischemic optic neuropathy and central retinal artery occlusion) have different funduscopic findings.
Classification criteria
The American College of Rheumatology has defined classification criteria for GCA (Table 2), but these criteria were developed for research purposes rather than for diagnosis. Temporal artery biopsies and MR angiograms are useful diagnostic adjuncts, but the diagnosis of GCA remains a clinical one.



Temporal artery biopsy and pathology
The decision to obtain a biopsy specimen should never delay the start of corticosteroids if clinical suspicion is high. The pathological features of an inflamed temporal artery may persist for at least 2 weeks after the start of corticosteroid treatment and probably considerably longer.4 The critical points are to begin corticosteroids promptly if justified by the clinical situation and to obtain a temporal artery biopsy specimen as soon thereafter as possible.
We usually recommend bilateral temporal artery biopsies. In 10% to 20% of biopsy-positive cases of GCA in which unilateral biopsies are obtained, the appropriate diagnosis is not made.5
The classic histopathology of GCA is transmural inflammation with mononuclear cells, varying degrees of concentric intimal proliferation, and internal elastic lamina fragmentation.6 However, the earliest histopathological change is a lymphoplasmacytic infiltrate in the adventitia. (Note that this infiltrate is not specific for GCA; it may occur in other forms of systemic vasculitis that involve the temporal arteries, such as microscopic polyangiitis.) Multinucleated giant cells are detected in at least 50% of GCA biopsy specimens but are not critical for the diagnosis (Figure 1).7
Figure 1 – A temporal artery biopsy of a patient with giant cell arteritis (GCA) shows a chronically inflamed blood vessel with granulomatous inflammation and intimal hyperplasia, which leads to significant luminal narrowing. Multinucleated giant cells are detected in half of GCA specimens but are not critical for the diagnosis.
Laboratory testing
The erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) level are elevated in most patients with GCA but to varying degrees. GCA is considered part of a small group of disorders (including osteomyelitis and multiple myeloma) that are associated with extreme elevations of the ESR. However, low to normal ESRs were reported in 10% of patients who had a new diagnosis of GCA in a population-based study.8
The ESR is an indirect measure of acute phase reactants and can be influenced by red blood cell size, hematocrit, and the presence of elevated serum immunoglobulins. The CRP level has the potential advantage of being a direct measure of acute phase reactants. However, there are no consistent data indicating that the CRP level reflects disease activity more accurately than does the ESR. In monitoring disease activity after the start of therapy, placing great emphasis on trends in either the ESR or CRP level often is a mistake. Acute phase reactant levels usually normalize swiftly after treatment is started. As prednisone is tapered, the ESR or CRP level may creep up to “above normal” for a particular laboratory. In the absence of clinical symptoms of GCA, such increases should not be considered a reflection of active disease and treatment should not be escalated.
Imaging
For some patients with GCA, imaging techniques have become increasingly important in making the diagnosis. However, they remain insufficiently refined for longitudinal assessment of disease activity. CT and MR and conventional angiography (Figure 2) are useful in assessing stenoses and aneurysms. These techniques also may detect wall thickening or, after contrast administration, enhancement; in some cases, enhancement corresponds to active inflammation.9,10


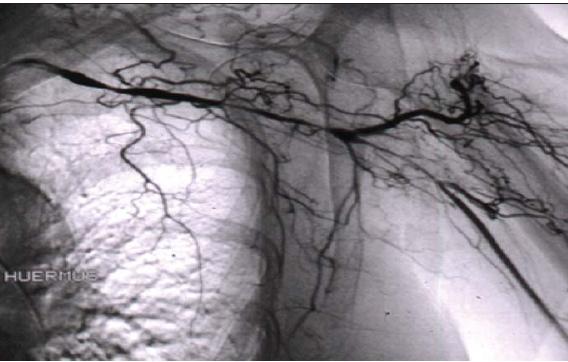
Figure 2 – An angiogram of the left upper extremity of a patient with giant cell arteritis shows serial smooth narrowings of the subclavian artery. This artery is essentially occluded in the area of the axilla, but the axillary artery is reconstituted by extensive collateral circulation.
Although cross-sectional imaging findings are quite helpful in making the original diagnosis, the correlation between areas of thickening or enhancement within the vessel wall and subsequent vascular narrowing is poor. This calls into question the reliability of these techniques in quantifying disease activity.
Substantial effort has been devoted to the use of Doppler ultrasonography in GCA. However, whether this modality is any more useful in making the diagnosis than a careful history and physical examination is unclear. In addition, the utility of ultrasonography diminishes substantially after corticosteroid therapy has begun.
Positron emission tomography (PET) scanning is an exciting imaging technique but expensive and still unproved for use in GCA. In a prospective study of 35 patients with GCA, contrast uptake was detected in the large vessels of 83% of them.11 Before broad acceptance of PET scanning in the clinic will be appropriate, further study is needed with a variety of control groups and patients with GCA in various stages of disease activity.
TREATMENT
Corticosteroids
Conventional approach. In most patients with GCA, the current standard approach to therapy is prednisone starting at a dosage of about 1 mg/kg/d and not exceeding 80 mg/d. Selection of the starting dosage is highly empirical, and clinicians’ practices vary. There also is no standard corticosteroid tapering regimen. We recommend the following guidelines:
• Use of 60 mg/d for 1 month is appropriate for initial suppression of disease activity.
• Tapering to 20 mg/d by the end of month 2 helps avoid excessive prednisone toxicity.
• A long “tail” to the prednisone taper is required, such that patients continue to take the medication for at least 1 year but spend most of that time taking less than 10 mg/d.
• Tapering in 1-mg decrements every 2 to 4 weeks (or even longer) at the end of the prednisone taper is appropriate.
Relapses are common in patients with GCA. About 50% experience a relapse during the first year of treatment, usually when their prednisone dosage goes below 10 mg/d.12,13 Disease flares require reinstitution of prednisone at a dosage that previously was effective in maintaining disease remission. Once the disease is controlled, a slower prednisone taper is recommended. Although many patients can discontinue prednisone in time, a few will require a low dosage of prednisone (5 to 10 mg/d) indefinitely to maintain disease quiescence.
All patients who take prolonged corticosteroid courses should receive 1.5 g/d of elemental calcium and 800 IU/d of vitamin D to prevent osteoporosis. Patients who take 7.5 mg/d or more of prednisone for more than 6 months also are candidates for bisphosphonate therapy. Bone mineral density studies are recommended at baseline and then every other year to monitor the development of osteoporosis.
Threatened or completed vision loss. Patients who present with threatened vision loss (eg, symptoms of amaurosis fugax or transient monocular blindness or other visual symptoms of concern) make up a special group among those who have GCA. They require the most aggressive possible treatment approach from the outset because vision loss in 1 or both eyes has disastrous implications.
We suggest hospitalizing these patients for administration of “pulse” methylprednisolone, 1 g/d for 3 days. Limited data suggest that if high-dose corticosteroids are used in this manner, patients who present with completed vision loss have a small chance of recovering some useful vision in the affected eye. In both threatened and completed vision loss, the use of low-dose aspirin is also suggested.
Investigational corticosteroid use. Given the numerous adverse effects of long-term corticosteroids, the use of shorter regimens and of corticosteroid-sparing agents is being explored for the management of GCA. In a randomized trial, 27 patients with GCA were assigned to 1 of 2 treatment arms: either a pulse methylprednisolone regimen (15 mg/kg [ideal body weight]/d for 3 days), followed by a standard tapering regimen, or intravenous saline plus prednisone 40 mg/d, followed by a prednisone taper identical to that of the other arm.14
Patients in the pulse arm tapered off prednisone more quickly and had a higher frequency of sustained remission after discontinuation of treatment at 1 year than did those in the other treatment group (78.5% vs 15.4%; P = .001). In addition, excluding the pulse methylprednisolone received at the start of therapy, patients in the pulse arm used less cumulative corticosteroid than did their counterparts in the other group. These interesting results suggest that a larger study of this approach to GCA treatment is appropriate.
Antiplatelet therapy
Two retrospective cohort studies have indicated clearly that low-dose aspirin should be a standard part of GCA therapy.15,16 Both studies demonstrated substantial reductions in odds ratios for vision loss and cerebral ischemic events.
One study reviewed 175 patients with GCA, 36 of whom were taking low-dose aspirin (100 mg/d) for cardiovascular disease before the diagnosis of GCA.16 All patients were treated with prednisone. The incidence of cranial ischemic events at presentation was significantly lower among patients taking aspirin at baseline (8% vs 29%; odds ratio, 0.22). In addition, among the 41 patients who began aspirin after their GCA diagnosis, cranial ischemic events were less common in the aspirin group (3% vs 13%). Similar benefits were noted in the other study; 60% of 143 patients with GCA were already receiving an antiplatelet agent (mostly aspirin) or warfarin at the time of their diagnosis.15 Based on the consistency of findings in these 2 studies and the somewhat low risk of adverse events, the use of low-dose aspirin (81 to 100 mg/d) in all patients who have GCA without contraindications appears to be prudent
Methotrexate
In a search for potential corticosteroid-sparing agents in the management of GCA, 2 double-blind placebo-controlled trials have evaluated the efficacy of methotrexate (MTX). In the smaller of the studies, patients given 10 mg/wk of MTX for 24 months in conjunction with standard corticosteroid therapy experienced significantly fewer relapses (45% vs 84.2%; P = .02) and used fewer cumulative corticosteroids than did those who used corticosteroids alone.12
In contrast, in a study that included twice the number of patients and used a higher dosage of MTX, the adjunctive use of MTX was not significantly better than the use of corticosteroids alone in the management of GCA.13 Patients who were treated with MTX plus prednisone did not have a significantly lower relapse rate than those treated with corticosteroids alone (57.5% vs 77.3%; P = .26). However, the MTX group did show a significantly lower rate of relapse that was heralded by PMR symptoms.
With such conflicting data, there is no uniform recommendation for the use of MTX in the management of GCA. Until additional data are available, we recommend the use of MTX only as a second-line agent in conjunction with corticosteroids and aspirin for patients who are refractory to or intolerant of conventional corticosteroid therapy.
Infliximab
Although case reports and small case series suggested that targeted tumor necrosis factor α (TNF-α) inhibition might be useful in GCA management, the results of a recent randomized controlled trial appear to debunk this idea.17 The results of this trial, combined with the negative results of TNF-α inhibition strategies in other forms of vasculitis,18 greatly diminish enthusiasm for the use of infliximab in GCA. Thus, to date, no therapeutic agent in GCA has been demonstrated to have a corticosteroid-sparing effect.
References:
References
- 1. Hunder GG. Epidemiology of giant-cell arteritis. Cleve Clin J Med. 2002;69(suppl 2):SII79- SII82.
- 2. Evans JM, O’Fallon WM, Hunder GG. Increased incidence of aortic aneurysm and dissection in giant cell (temporal) arteritis: a population-based study. Ann Intern Med. 1995;122:502-507.
- 3. Evans JM, Bowles CA, Bjornsson J, et al. Thoracic aortic aneurysm and rupture in giant cell arteritis: a descriptive study of 41 cases. Arthritis Rheum. 1994;37:1539-1547.
- 4. Achkar AA, Lie JT, Hunder GG, et al. How does previous corticosteroid treatment affect the biopsy findings in giant cell (temporal) arteritis? Ann Intern Med. 1994;120:987-992.
- 5. Seo P, Stone JH. Large-vessel vasculitis. Arthritis Rheum. 2004;51:128-139.
- 6. Albert DM, Searl SS, Craft JL. Histologic and ultrastructural characteristics of temporal arteritis: the value of the temporal artery biopsy. Ophthalmology. 1982;89:1111-1126.
- 7. Lie JT. Temporal artery biopsy diagnosis of giant cell arteritis: lessons from 1109 biopsies. Anat Pathol. 1996;1:69-97.
- 8. Salvarani C, Hunder GG. Giant cell arteritis with low erythrocyte sedimentation rate: frequency of occurrence in a population-based study. Arthritis Rheum. 2001;45:140-145.
- 9. Atalay MK, Bluemke DA. Magnetic resonance imaging of large vessel vasculitis. Curr Opin Rheumatol. 2001;13:41-47.
- 10. Stanson AW. Imaging findings in extracranial (giant cell) temporal arteritis. Clin Exp Rheumatol. 2000;18(4, suppl 20):S43-S48.
- 11. Blockmans D, de Ceuninck L, Vanderschueren S, et al. Repetitive 18F-fluorodeoxyglucose positron emission tomography in giant cell arteritis: a prospective study of 35 patients. Arthritis Rheum. 2006;55:131-137.
- 12. Jover JA, Hernandez-Garcia C, Morado IC, et al. Combined treatment of giant-cell arteritis with methotrexate and prednisone: a randomized, double-blind, placebo-controlled trial. Ann Intern Med. 2001;134:106-114.
- 13. Hoffman GS, Cid MC, Hellmann DB, et al. A multicenter, randomized, double-blind, placebo-controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum. 2002;46:1309-1318.
- 14. Mazlumzadeh M, Hunder GG, Easley KA, et al. Treatment of giant cell arteritis using induction therapy with high-dose glucocorticoids: a double-blind, placebo-controlled, randomized prospective clinical trial. Arthritis Rheum. 2006;54:3310-3318.
- 15. Lee MS, Smith SD, Galor A, Hoffman GS. Antiplatelet and anticoagulant therapy in patients with giant cell arteritis. Arthritis Rheum. 2006;54:3306-3309.
- 16. Nesher G, Berkun Y, Mates M, et al. Low-dose aspirin and prevention of cranial ischemic complications in giant cell arteritis. Arthritis Rheum. 2004;50:1332-1337.
- 17. Hoffman GS, Cid MC, Rendt-Zagar KE, et al. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med. 2007;146:621-630.
- 18. Wegener’s Granulomatosis Etanercept Trial (WGET) Research Group. Etanercept plus standard therapy for Wegener’s granulomatosis. N Engl J Med. 2005;352:35.














